
This post is the fourth in a series of five posts related to the paper “Melsted, Booeshaghi et al., Modular and efficient pre-processing of single-cell RNA-seq, bioRxiv, 2019“. The posts are:
- Near-optimal pre-processing of single-cell RNA-seq
- Single-cell RNA-seq for dummies
- How to solve an NP-complete problem in linear time
- Rotating the knee (plot) and related yoga
- High velocity RNA velocity
The “knee plot” is a standard single-cell RNA-seq quality control that is also used to determine a threshold for considering cells valid for analysis in an experiment. To make the plot, cells are ordered on the x-axis according to the number of distinct UMIs observed. The y-axis displays the number of distinct UMIs for each barcode (here barcodes are proxies for cells). The following example is from Aaron Lun’s DropletUtils vignette:

A single-cell RNA-seq knee plot.
High quality barcodes are located on the left hand side of the plot, and thresholding is performed by identifying the “knee” on the curve. On the right hand side, past the inflection point, are barcodes which have relatively low numbers of reads, and are therefore considered to have had failure in capture and to be too noisy for further analysis.
In Melsted, Booeshaghi et al., Modular and efficient pre-processing of single-cell RNA-seq, bioRxiv, 2019, we display a series of plots for a benchmark panel of 20 datasets, and the first plot in each panel (subplot A)is a knee plot. The following example is from an Arabidopsis thaliana dataset (Ryu et al., 2019; SRR8257100)

Careful examination of our plots shows that unlike the typical knee plot made for single-cell RNA-seq , ours has the x- and y- axes transposed. In our plot the x-axis displays the number of distinct UMI counts, and the y-axis corresponds to the barcodes, ordered from those with the most UMIs (bottom) to the least (top). The figure below shows both versions of a knee plot for the same data (the “standard” one in blue, our transposed plot in red):

Why bother transposing a plot?
We begin by observing that if one ranks barcodes according to the number of distinct UMIs associated with them (from highest to lowest), then the rank of a barcode with x distinct UMIs is given by f(x) where
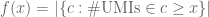
In other words, the rank of a barcode is interpretable as the size of a certain set. Now suppose that instead of only measurements of RNA molecules in cells, there is another measurement. This could be measurement of surface protein abundances (e.g. CITE-seq or REAP-seq), or measurements of sample tags from a multiplexing technology (e.g. ClickTags). The natural interpretation of #distinct UMIs as the independent variable and the rank of a barcode as the dependent variable is now clearly preferable. We can now define a bivariate function f(x,y) which informs on the number of barcodes with at least x RNA observations and y tag observations:

Nadia Volovich, with whom I’ve worked on this, has examined this function for the 8 sample species mixing experiment from Gehring et al. 2018. The function is shown below:

Here the x-axis corresponds to the #UMIs in a barcode, and the y-axis to the number of tags. The z-axis, or height of the surface, is the f(x,y) as defined above. Instead of thresholding on either #UMIs or #tags, this “3D knee plot” makes possible thresholding using both (note that the red curve shown above corresponds to one projection of this surface).
Separately from the issue described above, there is another subtle issue with the knee plot. The x-axis (dependent) variable really ought to display the number of molecules assayed rather than the number of distinct UMIs. In the notation of Melsted, Booeshaghi et al., 2019 (see also the blog post on single-cell RNA-seq for dummies), what is currently being plotted is |supp(I)|, instead of |I|. While |I| cannot be directly measured, it can be inferred (see the Supplementary Note of Melsted, Booeshaghi et al., 2019), where the cardinality of I is denoted by k (see also Grün et al,, 2014). If d denotes the number of distinct UMIs for a barcode and n the effective number of UMIs , then k can be estimated by
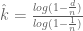
The function estimating k is monotonic so for the purpose of thresholding with the knee plot it doesn’t matter much whether the correction is applied, but it is worth noting that the correction can be applied without much difficulty.


Leave a comment
Comments feed for this article