
This post is the second in a series of five posts related to the paper “Melsted, Booeshaghi et al., Modular and efficient pre-processing of single-cell RNA-seq, bioRxiv, 2019“. The posts are:
- Near-optimal pre-processing of single-cell RNA-seq
- Single-cell RNA-seq for dummies
- How to solve an NP-complete problem in linear time
- Rotating the knee (plot) and related yoga
- High velocity RNA velocity

A few months ago, while working on the kallisto | bustools project, some of us in the lab were discussing various aspects of single-cell RNA-seq technology when the conversation veered into a debate over the meaning of some frequently used words and phrases in the art: “library complexity”, “library size”, “sensitivity”, “capture rate”, “saturation”, “number of UMIs”, “bork bork bork” etc. There was some sense of confusion. I felt like a dummy because even after working on RNA-seq for more than a decade, I was still lacking language and clarity about even the most basic concepts. This was perhaps not entirely my fault. Consider, for example, that the phrase “library size” is used to mean “the number of molecules in a cDNA library” by some authors, and the “number of reads sequenced” by others.
Since we were writing a paper on single-cell RNA-seq pre-processing that required some calculations related to the basic concepts (libraries, UMIs, and so on), we decided to write down notation for the key objects. After some back-and-forth, Sina Booeshaghi and I ended up drafting the diagram below that summarizes the sets of objects in a single-cell RNA-seq experiment, and the maps that relate them:

Structure of a single-cell RNA-seq experiment.
Each letter in this diagram is a set. The ensemble of RNA molecules contained within a single cell is denoted by R. To investigate R, a library (L) is constructed from the set of molecules captured from R (the set C). Typically, L is the result of of various fragmentation and amplification steps performed on C, meaning each element of C may be observed in L with some multiplicity. Thus, there is an inclusion map from C to L (arrow with curly tail), and an injection from C to R (arrows with head and tail). The library is interrogated via sequencing of some of the molecules in L, resulting in a set F of fragments. Subsequently, the set F is aligned or pseudoaligned to create a set B, which in our case is a BUS file. Not every fragment F is represented in B, hence the injection, rather than bijection, from B to F, and similarly from F to L. The set T consists of transcripts that correspond to molecules in C that were represented in B. Note that 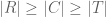
The diagram has three distinct parts. The sets on the top (L, F, B) are “lifted” from C and T by PCR. Without PCR one would be in an the ideal situation of measuring C directly to produce T, which would then be used to directly draw inferences about R. This is the hope for direct RNA sequencing, a technology that is promising but that cannot yet be applied at the scale of cDNA based methods. The sets U and I are intended to be seen as orthogonal to the rest of the objects. They relate to the UMIs which, in droplet single-cell RNA-seq technology, are delivered via beads. While the figure was designed to describe single-cell RNA-seq, it is quite general and possibly a useful model for many sequence census assays.
So what is all this formality good for? Nothing in this setup is novel; any practitioner working with single-cell RNA-seq already knows what the ingredients for the technology are. However I do think there is some trouble with the language and meaning of words, and hopefully having names and labels for the relevant sets can help in communication.
The questions
With some notation at hand, it is possible to precisely articulate some of the key technical questions associated with a single-cell RNA-seq experiment:
- The alignment (or pseudoalignment) problem: compute B from F.
- The pre-processing problem: what is the set T ?
- What is the library richness/complexity, i.e. what is |supp(L)|?
- What is the sensitivity, i.e. what is
?
- In droplet based experiments, what are the number of UMIs available to tag molecules in a cell, i.e. what is |U|?
These basic questions are sometimes confused with each other. For example, the capture rate refers to the proportion of cells from a sample that are captured in an experiment and should not be confused with sensitivity. The |supp(L)| is a concept that is natural to refer to when thinking about a cDNA library. Note that the “library size”, referred to in the beginning of this post, is used by molecular biologists to naturally mean |L|, and not |F| (this confusion was unfortunately disseminated by the highly influential RNA-seq papers Anders and Huber, 2010 and Robinson and Oshlack, 2010) . The support of another set, |supp(I)|, is one that is easy to measure but precisely because I is a multiset, 

This diagram in this post is just step 0 in discussing single-cell RNA-seq. There is a lot more subtlety and nuance in understanding and interpreting experiments (see Introduction to single-cell RNA-seq technologies). ∎

Leave a comment
Comments feed for this article