
This post is the first in a series of five posts related to the paper “Melsted, Booeshaghi et al., Modular and efficient pre-processing of single-cell RNA-seq, bioRxiv, 2019“. The posts are:
- Near-optimal pre-processing of single-cell RNA-seq
- Single-cell RNA-seq for dummies
- How to solve an NP-complete problem in linear time
- Rotating the knee (plot) and related yoga
- High velocity RNA velocity
During the past few years computational biologists have expended enormous effort in developing tools for processing and analyzing single-cell RNA-seq. This post describes yet another: the kallisto|bustools workflow for pre-processing single-cell RNA-seq. A preprint describing the method (Melsted, Booeshaghi et al. 2019) was recently posted on the bioRχiv.
 Number of single-cell RNA-seq tools (from the scRNA-tools catalog).
Number of single-cell RNA-seq tools (from the scRNA-tools catalog).
Given that there are so many programs, a natural question is: why on earth would we write yet another software program for generating a count matrix from single-cell RNA-seq reads when there are already plenty of programs out there? There’s alevin, cell ranger, dropseqpipe, dropseqtools, indrops… I’ve been going in alphabetical order but have to jump in with starsolo because it’s got the coolest name…now back to optimus, scruff, scpipe, scumi, umis, zumis, and I’m probably forgetting a few other something-umis. So why another one?
The answer requires briefly venturing back to a time long, long ago when RNA-seq was a fledgling, exciting new technology (~2009). At the time the notion of an “equivalence class” was introduced to the field (see e.g. Jiang and Wong, 2009 or Nicolae et al., 2011). Briefly, there is a natural equivalence relation on the set of reads in an RNA-seq experiment, where two reads are related when they are compatible with (i.e. could have originated from) exactly the same set of transcripts. The equivalence relation partitions the reads into equivalence classes, and, in a slight abuse of notation, the term “equivalence class” in RNA-seq is used to denote the set of transcripts corresponding to an equivalence class of reads. Starting with the pseudoalignment program kallisto that we published in Bray et al. 2016, it became possible to rapidly obtain the (transcript) equivalence classes for reads from an RNA-seq experiment.
When single-cell RNA-seq started to scale it became apparent to those of us working with equivalence classes for bulk RNA-seq that rather than counting genes from single-cell RNA-seq data, it would be better to examine what we called transcript compatibility counts (TCCs), i.e. counts of the equivalence classes (the origin of the term TCC is discussed in a previous blog post of mine). This vision has borne out: we recently published a paper demonstrating the power of TCCs for differential analysis of single-cell data (Ntranos, Yi et al. 2019) and I believe TCCs are ideal for many different single-cell RNA-seq analyses. So back to the question: why another single-cell RNA-seq pre-processing workflow?
Already in 2016 we wanted to be able to produce TCC matrices from single-cell RNA-seq data but there was no program to do it. My postdoc at the time, Vasilis Ntranos, developed a workflow, but in the course of working on a program he started to realize that there were numerous non-trivial aspects to processing single-cell RNA-seq. Even basic questions, such as how to correct barcodes or collapse UMIs required careful thought and analysis. As more and more programs for single-cell RNA-seq pre-processing started to appear, we examined them carefully and noted two things: 1. Most were not able to output TCC matrices and 2. They were, for the most part, based on ad hoc heuristics and unvalidated methods. Many of the programs were not even released with a preprint or paper. So we started working on the problem.
A key insight was that we needed a new format to allow for modular pre-processing. So we developed such a format, which we called the Barcode, UMI, Set (BUS) format, and we published a paper about it earlier this year (Melsted, Ntranos et al., 2019). This allowed us to start investigating different algorithms for the key steps, and to rearrange them and plug them in to an overall workflow as needed. Finally, after careful consideration of each of the key steps, weighing tradeoffs between efficiency and accuracy, and extensive experimentation, we settled on a workflow that is faster than any other method and based on reason rather than intuition. The workflow uses two programs, kallisto and bustools, and we call it the kallisto|bustools workflow. Some highlights:
- kallisto|bustools can produce a TCC matrix. The matrix is compatible with the gene count matrix (it collapses to the latter), and of course gene count matrices can be output as well for use in existing downstream tools.
- The workflow is very very fast. With kallisto|bustools very large datasets can be processed in minutes. The title of this post refers to the workflow as “near-optimal” because it runs in time similar to the unix word count function. Maybe it’s possible to be a bit faster with some optimizations, but probably not by much:
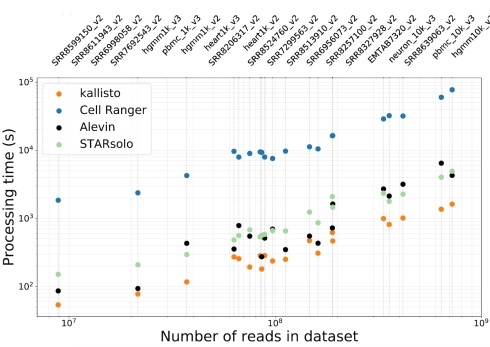
- kallisto|bustools uses very little memory. We worked hard to achieve this feature, as we wanted it to be useful for large-scale analyses that are going to be performed by consortia such as the Human Cell Atlas project. The workflow currently uses ~3.5Gb of RAM for processing 10x v2 chemistry data, and ~11Gb RAM for 10x v3 chemistry data; both numbers are independent of the number of reads being processed. This means users can pre-process data on a laptop:

- The workflow is modular, thanks to its reliance on the flexible BUS format. It was straightforward to develop an RNA velocity workflow (more on this in a companion blog post). It will be easy to adapt the workflow to various technologies, to multiomics experiments, and to any custom analysis required:

- We tried to create comprehensive, yet succinct documentation to help make it easy to use the software (recommendations for improvements are welcome). We have online tutorials, as well as videos for novices:
– Installation instructions (and video)
– Getting started tutorial (and video).
– Manuals for kallisto and bustools.
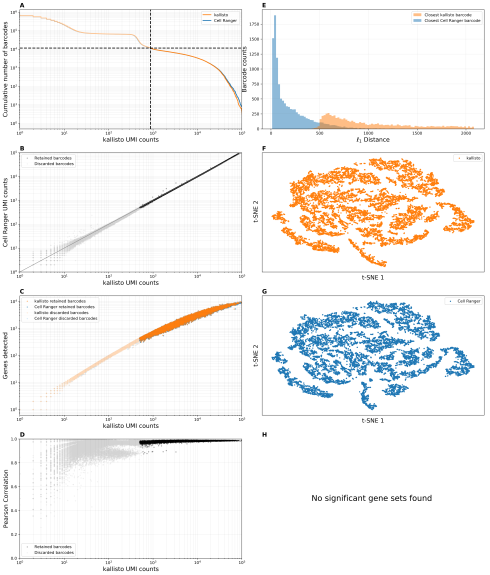
– Complete code for reproducing all the results in the preprint - We were not lazy. In our tests we found variability in performance on different datasets so we tested the program extensively and ran numerous benchmarks on 10x Genomics data to validate Cell Ranger with respect to kallisto|bustools (note that Cell Ranger’s methods have been neither validated nor published). We compiled a benchmark panel consisting of 20 datasets from a wide variety of species. This resulted in 20 supplementary figures, each with 8 panels showing: a) the number of genes detected, b) concordance in counts per gene, c) number of genes detected, d) correlation in gene counts by cell, e) spatial separation between corresponding cells vs. neighboring cells, f,g) t-SNE analysis, h) gene set analysis to detect systematic differences in gene abundance estimation (see example below for the dataset SRR8257100 from the paper Ryu et al., 2019). We also examined in detail results on a species mixing experiment, and confirmed that Cell Ranger is consistent with kallisto on that as well. One thing we did not do in this paper is describe workflows for different technologies but such workflows and accompanying tutorials will be available soon:
- In addition we ran a detailed analysis on the 10x Genomics 10k E18 mouse brain dataset to investigate whether Cell Ranger pre-processing produces different results than kallisto insofar as downstream analyses are concerned. We looked at dimensionality reduction, clustering, identification of marker genes, marker gene prevalence, and pseudotime. The results were all highly concordant. An example (the pseudotime analysis) is shown below:

- We did the math on some of the basic aspects of single-cell RNA-seq. We’re not the first to do this (see, e.g. Petukhov et al., 2018), but one new result we have is an estimation of the UMI diversity on beads. This should be useful for those developing new technologies, or trying to optimize existing protocols:

Note that this post is the first in a series of five that discuss in more detail various aspects of the paper (see links at the top). Finally, a note on reproducibility and usability:
The development of the kallisto|bustools workflow, research into the methods, compilation of the results, and execution of the project required a tremendous team effort, and in working on it I was thinking of the first bioinformatics tool I wrote about and posted to the arXiv (the bioRxiv didn’t exist yet). The paper was:
Nicolas Bray and Lior Pachter, MAVID: Constrained ancestral alignment of multiple sequences, arXiv, 2003.
At the time we posted the code on our own website (now defunct, but accessible via the Wayback machine). We did our best to make the results reproducible but we were limited in our options with the tools available at the time. Furthermore, posting the preprint was highly unusual; there was almost no biology preprinting at the time. Other things have stayed the same. Considerations of software portability, efficiency and documentation were relevant then and remain relevant now.
Still, there has been an incredible development in the tools and techniques available for reproducibility and usability since that time. A lot of the innovation has been made possible by cloud infrastructure, but much of the development has been the result of changes in community standards and requirements (see e.g., Weber et al., 2019). I thought I’d compile a list of the parts and pieces of the project; they are typical for what is needed for a bioinformatics project today and comparing them to the bar in 2003 is mind boggling:
Software: GitHub repositories (kallisto and bustools); releases of binaries for multiple operating systems (Mac, Linux, Windows, Rock64); portable source code with minimal dependencies; multithreading; memory optimization; user interface.
Paper: Preprint (along with extensive Supplement providing backup for every result and claim in the main text); GitHub repository with code to reproduce all the figures/results in the preprint (reproducibility code includes R markdown, python notebooks, snakemake, software versions for every program used, fixed seeds).
Documentation: Manuals for the software; Tutorials for learning to use the code; Explanatory videos (all required materials posted on Github or available on stable websites for download).
The totality of work required to do all of this was substantial. Páll Melsted was the primary developer of kallisto and he wrote and designed bustools, which has been the foundation of the project. The key insight to adopt the BUS format was work in collaboration with Vasilis Ntranos. This was followed by long conversations on the fundamentals of single-cell RNA-seq with Jase Gehring. Sina Booeshaghi carried the project. He was responsible for the crucial UMI collapsing analysis, and put together the paper. Fan Gao, director of the Caltech Bioinformatics Resource Center, set up and implemented the extensive benchmarking, and helped fine-tune the algorithms and converge to the final approach taken. Lambda Lu conducted what I believe to be the most in-depth and detailed analysis to date of the effect of pre-processing on results. Her framework should serve as a template for future development work in this area. Eduardo Beltrame designed the benchmark panels and had a key insight about how to present results that is described in more detail in the companion post on rotating the knee plot. He also helped in the complex task of designing and building the companion websites for the project. Kristján Eldjarn Hjörleifsson helped with the RNA velocity work and helped make custom indices that turned out to be fundamental in understanding the performance of pseudoalignment in the single-cell RNA-seq setting. Sina Booeshaghi spent a lot of time thinking about how to optimize the user experience, making the tutorials and videos, and working overall to make the results of the paper not just reproducible, but the the methods usable.


Leave a comment
Comments feed for this article