
The development of microarray technology two decades ago heralded genome-wide comparative studies of gene expression in human, but it was the widespread adoption of RNA-Seq that has led to differential expression analysis becoming a staple of molecular biology studies. RNA-Seq provides measurements of transcript abundance, making possible not only gene-level analyses, but also differential analysis of isoforms of genes. As such, its use has necessitated refinements of the term “differential expression”, and new terms such as “differential transcript expression” have emerged alongside “differential gene expression”. A difficulty with these concepts is that they are used to describe biology, statistical hypotheses, and sometimes to describe types of methods. The aims of this post are to provide a unifying framework for thinking about the various concepts, to clarify their meaning, and to describe connections between them.
To illustrate the different concepts associated to differential expression, I’ll use the following example, consisting of a comparison of a single two-isoform gene in two conditions (the figure is Supplementary Figure 1 in Ntranos, Yi et al. Identification of transcriptional signatures for cell types from single-cell RNA-Seq, 2018):

The isoforms are labeled primary and secondary, and the two conditions are called “A” and “B”. The black dots labeled conditions A and B have x-coordinates 



Biology
Below is a list of terms used to characterize changes in expression:
Differential transcript expression (DTE) is change in one of the isoforms. In the figure, this is represented (conceptually) by the two red lines along the x- and y-axes respectively. Algebraically, one might compute the change in the primary isoform by 

Differential gene expression (DGE) is the change in the overall output of the gene. Change in the overall output of a gene is change in the direction of the line 





Differential transcript usage (DTU) is the change in relative expression between the primary and secondary isoforms. This can be interpreted geometrically as the angle between the two points, or alternatively as the length (as given by some norm) of the green line labeled DTU. As with DTE and DGE, DTU is also a term used to describe a certain kind of difference in expression between two conditions, one in which the line joining the two points has a slope of -1. DTU events are most likely controlled by post-transcriptional regulation.
Gene differential expression (GDE) is represented by the red line. It is the amount of change in expression along in the direction of line joining the two points. GDE is a notion that, for reasons explained below, is not typically tested for, and there are few methods that consider it. However GDE is biologically meaningful, in that it generalizes the notions of DGE, DTU and DTE, allowing for change in any direction. A gene that exhibits some change in expression between conditions is GDE regardless of the direction of change. GDE can represent complex changes in expression driven by a combination of transcriptional and post-transcriptional regulation. Note that DGE, DTU and DTE are all special cases of GDE.
If the 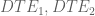

Statistics
The terms DTE, DGE, DTU and GDE have an intuitive biological meaning, but they are also used in genomics as descriptors of certain null hypotheses for statistical testing of differential expression.
The differential transcript expression (DTE) null hypothesis for an isoform is that it did not change between conditions, i.e. 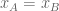
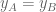
The differential gene expresión (DGE) null hypothesis is that there is no change in overall expression of the gene, i.e. 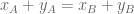
The differential transcript usage (DTU) null hypothesis is that there is no change in the difference in expression of isoforms, i.e. 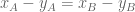
The gene differential expression (GDE) null hypothesis is that there is no change in expression in any direction, i.e. for all constants 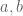

The union differential transcript expression (UDTE) null hypothesis is that there is no change in expression of any isoform. That is, that 

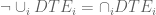
Not that 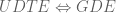
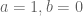

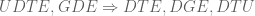
This is clear because if 

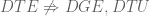
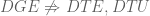

Methods
The terms DGE, DTE, DTU and GDE also used to describe methods for differential analysis.
A differential gene expression method is one whose goal is to identify changes in overall gene expression. Because DGE depends on the projection of the points (representing gene abundances) to the line y=x, DGE methods typically take as input gene counts or abundances computed by summing transcript abundances
A differential transcript expression method is one whose goal is to identify individual transcripts that have undergone DTE. Early methods for DTE were Cufflinks (now Cuffdiff2) and MISO, and more recently sleuth, which improves DTE accuracy by modeling uncertainty in transcript quantifications. A key issue with DTE is that there are many more transcripts than genes, so that rejecting DTE null hypotheses is harder than rejecting DGE null hypotheses. On the other hand, DTE provides differential analysis at the highest resolution possible, pinpointing specific isoforms that change and opening a window to study post-transcriptional regulation. A number of recent examples highlight the importance of DTE in biomedicine (see, e.g., Vitting-Seerup and Sandelin 2017). Unfortunately DTE results do not always translate to testable hypotheses, as it is difficult to knock out individual isoforms of genes.
A differential transcript usage method is one whose goal is to identify genes whose overall expression is constant, but where isoform switching leads to changes in relative isoform abundances. Cufflinks implemented a DTU test using Jensen-Shannon divergence, and more recently RATs is a method specialized for DTU.
As discussed in the previous section, none of null hypotheses DGE, DTE and DTU imply any other, so users have to choose, prior to performing an analysis, which type of test they will perform. There are differing opinions on the “right” approach to choosing between DGE, DTU and DTE. Sonseson et al. 2016 suggest that while DTE and DTU may be appropriate in certain niche applications, generally it’s better to choose DGE, and they therefore advise not to bother with transcript-level analysis. In Trapnell et al. 2010, an argument was made for focusing on DTE and DTU, with the conclusion to the paper speculating that “differential RNA level isoform regulation…suggests functional specialization of the isoforms in many genes.” Van den Berge et al. 2017 advocate for a middle ground: performing a gene-level analysis but saving some “FDR budget” for identifying DTE in genes for which the UDTE null hypothesis has been rejected.
There are two alternatives that have been proposed to get around the difficulty of having to choose, prior to analysis, whether to perform DGE, DTU or DTE:
A differential transcript expression aggregation (DTE->G) method is a method that first performs DTE on all isoforms of every gene, and then aggregates the resulting p-values (by gene) to obtain gene-level p-values. The “aggregation” relies on the observation that under the null hypothesis, p-values are uniformly distributed. There are a number of different tests (e.g. Fisher’s method) for testing whether (independent) p-values are uniformly distributed. Applying such tests to isoform p-values per gene provides gene-level p-values and the ability to reject UDTE. A DTE->G method was tested in Soneson et al. 2016 (based on Šidák aggregation) and the stageR method (Van den Berge et al. 2017) uses the same method as a first step. Unfortunately, naïve DTE->G methods perform poorly when genes change by DGE, as shown in Yi et al. 2017. The same paper shows that Lancaster aggregation is a DTE->G method that achieves the best of both the DGE and DTU worlds. One major drawback of DTE->G methods is that they are non-constructive, i.e. the rejection of UDTE by a DTE->G method provides no information about which transcripts were differential and how. The stageR method averts this problem but requires sacrificing some power to reject UDTE in favor of the interpretability provided by subsequent DTE.
A gene differential expression method is a method for gene-level analysis that tests for differences in the direction of change identified between conditions. For a GDE method to be successful, it must be able to identify the direction of change, and that is not possible with bulk RNA-Seq data. This is because of the one in ten rule that states that approximately one predictive variable can be estimated from ten events. In bulk RNA-Seq, the number of replicates in standard experiments is three, and the number of isoforms in multi-isoform genes is at least two, and sometimes much more than that.
In Ntranos, Yi et al. 2018, it is shown that single-cell RNA-Seq provides enough “replicates” in the form of cells, that logistic regression can be used to predict condition based on expression, effectively identifying the direction of change. As such, it provides an alternative to DTE->G for rejecting UDTE. The Ntranos and Yi GDE methods is extremely powerful: by identifying the direction of change it is a DGE methods when the change is DGE, it is a DTU method when the change is DTU, and it is a DTE method when the change is DTE. Interpretability is provided in the prediction step: it is the estimated direction of change.
Remarks
The discussion in this post is based on an example consisting of a gene with two isoforms, however the concepts discussed are easy to generalize to multi-isoform genes with more than two transcripts. I have not discussed differential exon usage (DEU), which is the focus of the DEXSeq method because of the complexities arising in genes which don’t have well-defined shared exons. Nevertheless, the DEXSeq approach to rejecting UDTE is similar to DTE->G, with DTE replaced by DEU. There are many programs for DTE, DTU and (especially) DGE that I haven’t mentioned; the ones cited are intended merely to serve as illustrative examples. This is not a comprehensive review of RNA-Seq differential expression methods.
Acknowledgments
The blog post was motivated by questions of Charlotte Soneson and Mark Robinson arising from an initial draft of the Ntranos, Yi et al. 2018 paper. The exposition was developed with Vasilis Ntranos and Lynn Yi. Valentine Svensson provided valuable comments and feedback.

13 comments
Comments feed for this article
February 15, 2018 at 8:47 am
Eduardo Eyras
Thanks for the post Lior.
I was wondering whether the null hypothesis for DTU should be that the rate of the two abundances is constant x_A / y_A = x_B / y_B, in this way, regardless of the gene expression, the isoforms occur with the same proportion. A gene expression change DGE without DTU would then occur along lines orthogonal to the DTU lines.
We drew some DTU lines in an earlier work about isoform switches in cancer: https://www.ncbi.nlm.nih.gov/pubmed/25578962
Here we showed that you could also define DTU as reversals in the ranking of isoform abundances.
February 15, 2018 at 9:25 am
Lior Pachter
Yes- you’re absolutely right that there is an alternative way to think about DTU. I allude to it in the section on DTU (“This can be interpreted geometrically as the angle between the two points”). The tangent of this angle is just the ratio you mention, and the magnitude changes along lines orthogonal to the DTU *circles*. I originally thought of introducing the polar coordinate viewpoint in more detail in the post, but decided at the end to stick with Cartesian coordinates for simplicity. One thought that comes to mind in light of your remark, is that the polar definition of DTU is discordant with the cartesian definition of DGE. The latter (which is in the blog post) is typically the one underlying DGE methods, however DGE in that sense can include DTU in the polar sense. Perhaps one way to resolve this problem is to think more in terms of GDE.
February 15, 2018 at 11:14 am
Donald Forsdyke
IT BEGAN IN THE 1980s
You write that: “The development of microarray technology two decades ago heralded genome-wide comparative studies of gene expression,” so now “differential expression analysis” is “a staple of molecular biology studies.”
Actually, genome-wide comparative studies involving differential RNA expression became feasible in the early 1980s. We stimulated cultured human lymphocytes to begin proliferating and two hours later isolated RNA from stimulated and control cultures. The comparison of cDNA clones lead to the specific identification of newly transcribed RNAs. The novel genes we unearthed included chemokines (CCL3), transcription factors (EGR1), a regulator of G-protein signalling (RGS2), a powerful inhibitor of triglyceride lipase (G0S2), an enzyme salvaging NAD (NAMPT), a regulator of mRNA lifespan (TTP), and various oncogenes (e.g. FosB).
At that time, folks were more interested in surface receptors that were bound by ligands (e.g. the T cell receptor), rather than the genes that were activated to transcribe when a receptor was activated. But studies of each of these genes, and classes of genes, are now well funded.
February 15, 2018 at 11:39 am
Lior Pachter
That’s a very good point. Thanks for catching my human-centric fallacy. I’ve updated the post to be more accurate.
February 15, 2018 at 5:31 pm
forsdyke
Another human-centric fallacy to which peer-reviewers are prone, is to dismiss differential expression analysis as a “fishing expedition.” Should you come across this, it might be helpful to point out that the wise fisherman knows both where to cast his nets, and at what time of day.
February 15, 2018 at 2:59 pm
Damian Kao
Thanks for this post Lior. This is a really cool way to think/visualize about DE.
It looks like GDE is a good holistic metric that assessed DE based on both DGE and DTU. Is it possible to calculate the contribution of DGE/DTU to GDE?
February 16, 2018 at 3:31 am
Nicolas Rosewick
Hi Lior,
I found a small mistake in Suppl. figure 2. The last two panels are wrongly labelled: There is two “d” . Should be e. and f. ; not d. and e. for the two last panels.
Beside that nice paper !
March 11, 2018 at 9:05 pm
Sahin Naqvi
Hi Lior, thanks for the post.
Just a small comment, when you say “DGE can be understood to be the result of transcriptional regulation, driving overall gene expression up or down,” I don’t think that’s strictly true; in addition to transcriptional regulation, post-transcriptional regulation of many flavors can also change overall gene expression levels, independently of or concordant with changes in transcript usage. It’s certainly reasonable to assume that in many cases, transcriptional regulation is the primary driver of DGE, but it doesn’t have to be the only one.
March 11, 2018 at 9:18 pm
Lior Pachter
Your point is well taken. I agree.
March 23, 2018 at 2:17 pm
john
Good post! What about DEE (diff exon expression)?
July 18, 2020 at 12:05 pm
Callie Burt
This is super helpful, thank you for writing.
Perhaps I missing something obvious, but by measuring gene expression (conceptualized, as I understand as transcript production) as current transcript abundance, are we assuming that different degradation rates of mRNA is unimportant or random, even though there is evidence it isn’t (e.g., Yang et al. 2003)? Or perhaps the obvious is that we are looking at relative changes (differential) versus overall gene expression (as transcript production).
Should (how, can, does) anyone account for different mRNA decay rates when looking at transcript abundance to capture ‘gene expression’ as overall mRNA transcripts produced from a given gene?
(Caveat, this question is from a social scientist now training in genomics who admits to significant knowledge limitations, which I am trying to address.)
July 19, 2020 at 2:07 pm
Lior Pachter
You’re zeroing in on several key points and limitations of transcriptome measurements.
– transcript abundance measurements (specifically polyadenylated mRNA that is typically measured in most bulk or single-cell RNA-seq) is not the same as protein abundance.
– Degradation rates are uneven and this is especially crucial when making inferences about dynamics (e.g. as is done with RNA velocity in single-cell RNA-seq).
– DGE, GDE or DTU as discussed in this blog post operates under a variety of assumptions. In addition to the ones you highlighted are also mundane technical issues (just as an example, without unique molecular identifiers bulk RNA-seq measurements may be confounded by PCR biases).
On top of all of this many experiments are performed with (too) few replicates.
More optimistically, I think it’s possible to conclude from the large number of experiments that have been undertaken, that differential analysis of RNA-seq does reveal transcripts that for the most part are consistent with known biology, and therefore discoveries can and have been made this way, especially looking at transcripts with large effects. Magnitude of effect is an important complement to statistical significance; I view this as so central to the endeavor that I chose it as one of the first topics to write about when I started this blog (https://liorpachter.wordpress.com/2013/08/26/magnitude-of-effect-vs-statistical-significance/).
July 20, 2020 at 3:13 pm
Callie Burt
Many thanks for your response and clarification! Moving to your earlier post now (sorry I missed!).