
RNA-Seq is the new kid on the block, but there is still something to be learned from the stodgy microarray. One of the lessons is hidden in a tech report by Daniela Witten and Robert Tibshirani from 2007: “A comparison of fold-change and the t-statistic for microarray data analysis“.
The tech report makes three main points. The first is that it is preferable to use a modified t-statistic rather than the ordinary t-statistic. This means that rather than comparing (normalized) means using
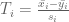
where 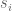
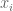
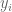

where 

The second point made is that the intuition that reproducibility implies accuracy is not correct (fold change had been proposed for use instead of a t-statistic because the results were more reproducible).
The third point, in my opinion the most important one, I quote directly from the report:
“A researcher should choose the measure of differential expression based on the biological system of interest. If large absolute changes in expression are relevant to the system, then fold-change should be used; on the other hand, if changes in expression relative to the underlying noise are important, then a modified t-statistic is preferable.”
How does this pertain to RNA-Seq? Microarray experiments and RNA-Seq both measure expression but the translation of methods for the analysis of one platform to the other can be non-trivial. One reason is that in RNA-Seq experiments accurately measuring “fold-change” is difficult. Read counts accumulated across a gene cannot be used directly to estimate fold change because the transcripts making up the gene may have different lengths. For this reason, methods such as Cufflinks, RSEM or eXpress (and most recently Sailfish recently reviewed on this blog) use the EM algorithm to “deconvolute” ambiguously mapped reads. The following thought experiment (Figure 1 in our paper describing Cufflinks/Cuffdiff 2) illustrates the issue:

Changes in fragment counts for a gene do not necessarily equal a change in expression. The “exon-union” method counts reads falling on any of a gene’s exons, whereas the “exon-intersection” method counts only reads
on constitutive exons. Both of the exon-union and exon-intersection counting schemes may incorrectly estimate a change in expression in genes with multiple isoforms as shown in the table. It is important to note that the problem of fragment assignment described here in the context of RNA-Seq is crucial for accurate estimation of parameters in many other *Seq assays.
“Count-based” methods for differential expression, such as DESeq, work directly with accumulated gene counts and are based on the premise that even if estimated fold-change is wrong, statistical significance can be assessed based on differences between replicates. In our recent paper describing Cuffdiff 2 (with a new method for differential abundance analysis) we examine DESeq (as a proxy for count-based methods) carefully and show using both simulation and real data that fold-change is not estimated accurately. In fact, even when DESeq and Cufflinks both deem a gene to be differentially expressed, and even when the effect is in the same direction (e.g. up-regulation), DESeq can (and many times does) estimate fold-change incorrectly. This problem is not specific to DESeq. All “count based” methods that employ naive heuristics for computing fold change will produce inaccurate estimates:

Comparison of fold-change estimated by Cufflinks (tail of arrows) vs. “intersection-count” (head of arrows) reproduced from Figure 5 of the supplementary material of the Cuffdiff 2 paper. “Intersection-count” consists of the accumulated read counts in the regions shared among transcripts in a gene. The x-axis shows array fold change vs. the estimated fold-change on the y-axis. For more details on the experiment see the Cuffdiff 2 paper.
In other words,
it is essential to perform fragment assignment in a biological context where absolute expression differences are relevant to the system.
What might that biological context be? This is a subjective question but in my experience users of microarrays or RNA-Seq (including myself) always examine fold-change in addition to p-values obtained from (modified) t-statistics or other model based statistics because the raw fold-change is more directly connected to the data from the experiment.
In many settings though, statistical significance remains the gold standard for discovery. In the recent epic “On the immortality of television sets: ‘function’ in the human genome according to the evolution-free gospel of ENCODE“, Dan Graur criticizes the ENCODE project for reaching an “absurd conclusion” through various means, among them the emphasis of “statistical significance rather than magnitude of effect”. Or, to paraphrase Samuel Johnson,
statistical significance is the last refuge from a poor analysis of data.

6 comments
Comments feed for this article
September 13, 2013 at 11:51 am
Jonathan Jacobs (@bioinformer)
Great commentary here. It’s good to see the debate of count methods vs. FKPM methods still going.
I think your analysis method of choice also depends on the application and/or desired goal of the project. For time course experiments – I’ve found FPKM methods to work the best. For example: we’re setting up to carry out an RNA-Seq time course analysis of a series with relatively low count data (5M -10M reads per sample, triplicate biological replicates, four conditions (mock vs. infected with one of 3 virus isolates), and four time points, and for both miRNA and mRNA (polyA). It’s over 100 RNA-Seq samples altogether (hence the overall low read count per sample), so we’re using ERCC spike in controls and normalizing between samples by fitting to the ERCC data.
Again – thanks. Glad to have found your blog.
October 22, 2013 at 1:34 am
Jari Niemi
Here I just want to highlight a real well known biological example regarding the “Changes in fragment counts for a gene do not necessarily equal a change in expression” which is illustrated in Figure 1.
Here is an article which is a nice real biological example:
Z. Guo, et al. “A New Trick of an Old Molecule: Androgen Receptor Splice Variants Taking the Stage?!”, Int J Biol Sci. 2011; 7(6): 815–822. (URL: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3133889/ )
Shortly, AR gene is the one of the key players in prostate cancer and for some reason the tumor cells need AR gene. When the prostate tumor cells are treated with drugs which inhibit AR gene after a while the prostate tumor cells become resistant to inhibition of AR gene by using alternative splicing. Basically the AR gene becomes much “shorter” (the last exons are not expressed anymore) in resistant prostate tumor cells because the today’s drugs which inhibit AR are binding exactly to those last exons which are not expressed anymore. Therefore the drug cannot bind anymore to AR and it fails to inhibit it and the prostate tumor cells become “resistant”.
It would be interesting to do a DEG genes for this setup and see what tools/methods find the AR gene here DEG. One would expect from biological point of view that the AR’s gene expression is the same in both conditions (before and after that treatment with AR inhbitors). I guess that the methods which use “raw-counts” would find AR gene as DEG and Cuffdiff as not DEG.
October 22, 2013 at 1:40 am
Jari Niemi
And another article more recent about AR alternative splicing and AR inhibition is:
Y. Li et al. “Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines.”, Cancer Res. 2013 Jan 15;73(2):483-9.
http://www.ncbi.nlm.nih.gov/pubmed/23117885
March 18, 2014 at 5:50 pm
Anon
God knows I’ve been guilty of this before as well (trumpeting statistical significance while not examining magnitude of effect) and will have to work extra hard in the future to avoid this. I wonder if this problem is also what lead to a general pessimism about GWAS. Open up any Nature Genetics paper and you’ll see Manhattan plots showing p-values. The amazing p-values, however, belie the fact that risk-ratio or odds-ratio for any given SNP are often very close to 1.
May 27, 2015 at 12:17 am
Harry Hab
And don’t get me started on power analysis…
August 17, 2015 at 2:17 pm
A sleuth for RNA-Seq | Bits of DNA
[…] gene abundances by adding up counts to their genomic region. One consequence of this, is that it is not possible to accurately measure fold change between conditions by using counts to gene feat…. In other words, count-based methods are problematic even at the gene-level and it is necessary to […]