
Last Monday some biostatisticians/epidemiologists from Australia published a paper about a “visualization tool which may allow greater understanding of medical and epidemiological data”:
H. Wand et al., “Quilt Plots: A Simple Tool for the Visualisation of Large Epidemiological Data“, PLoS ONE 9(1): e85047.
A brief look at the “paper” reveals that the quilt plot they propose is a special case of what is commonly known as a heat map, a point the authors acknowledge, with the caveat that they claim that
” ‘heat maps’ require the specification of 21 arguments including hierarchical clustering, weights for reordering the row and columns dendrogram, which are not always easily understood unless one has an extensive programming knowledge and skills. One of the aims of our paper is to present ‘‘quilt plots’’ as a useful tool with simply formulated R-functions that can be easily understood by researchers from different scientific backgrounds without high-level programming skills.”
In other words, the quilt plot is a simplified heat map and the authors think it should be used because specifying parameters for a heat map (in R) would require a terrifying skill known as programming. This is of course all completely ridiculous. Not only does usage of R not require programming skill, there are also simplified heat map functions in many programming languages/computation environments that are as simple as the quilt plot function.
The fact that a paper like this was published in a journal is preposterous, and indeed the authors and editor of the paper have been ridiculed on social media, blogs and in comments to their paper on the PLoS One website.
BUT…
Wand et al. do have one point… those 21 parameters are not an entirely trivial matter. In fact, the majority of computational biologists (including many who have been ridiculing Wand) appear not to understand heat maps themselves, despite repeatedly (ab)using them in their own work.
What are heat maps?
In the simplest case, heat maps are just the conversion of a table of numbers into a grid with colored squares, where the colors represent the magnitude of the numbers. In the quilt plot paper that is the type of heat map considered. However in applications such as gene expression analysis, heat maps are used to visualize distances between experiments.
Heat maps have been popular for visualizing multiple gene expression datasets since the publication of the “Eisengram” (or the guilt plot?). So when my student Lorian Schaeffer and I recently needed to create a heat map from RNA-Seq abundance estimates in multiple samples we are analyzing with Ryan Forster and Dirk Hockemeyer, we assumed there would be a standard method (and software) we could use. However when starting to look at the literature we quickly found 3 papers with 4 different opinions about which similarity measure to use:
- In “The evolution of gene expression levels in mammalian organs“, Brawand et al., Nature 478 (2011), 343-348, the authors use two different distance measures for evaluating the similarity between samples. They use the measure
where
is Spearman’s rank correlation (Supplementary figure S2) and also the Euclidean distance (Supplementary figure S3).
- In “Differential expression analysis for sequence count data“, Anders and Huber, Genome Biology 11 (2010), R106, a heat map tool is provided that is based on Euclidean distance but differs from the method in Brawand et al. by first performing variance stabilization on the abundance estimates.
- In “Evolutionary Dynamics of Gene and Isoform Regulation in Mammalian Tissues“, Merkin et al., Science 338 (2012), a heat map is displayed based on the Jensen-Shannon metric.
There are also the folks who don’t worry too much and just try anything and everything (for example using the heatmap.2 function in R) hoping that some distance measure produces the figure they need for their paper. There are certainly a plethora of distance measures for them to try out. And even if none of the distance measures provide the needed figure, there is always the opportunity to play with the colors and shading to “highlight” the desired result. In other words, heat maps are great for cheating with what appears to be statistics.
We wondered… what is the “right” way to make a heat map?
Consider first the obvious choice for measuring similarity: Euclidean distance. Suppose that we are estimating the distance between abundance estimates from two RNA-Seq experiments, where for simplicity we assume that there are only three transcripts (A,B,C). The two abundance estimates can be represented by 3-tuples 
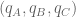

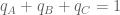








The Jensen-Shannon divergence, defined for two distributions 
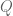
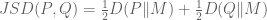
where 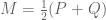
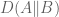


The 2-simplex with contour lines showing points equidistant
from the probability distribution (1/3, 1/3, 1/3) for the JSD metric.
The meaning of the JSD metric is not immediately apparent based on its definition, but a number of results provide some insight. First, the JSD metric can be approximated by Pearson’s 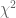
In “Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation“, Trapnell et al., Nature Biotechnology 28 (2010), we used the JSD metric to examine changes to relative isoform abundances in genes (see, for example, the Minard plot in Figure 2c). This application of the JSD metric makes sense, however the JSD metric is not a panacea. Consider Figure 1 in the Merkin et al. paper mentioned above. It displays a heat map generated from 7713 genes (genes with singleton orthologs in the five species studied). Some of these genes will have higher expression, and therefore higher variance, than others. The nature of the JSD metric is such that those genes will dominate the distance function, so that the heat map is effectively generated only from the highly abundant genes. Since there is typically an (approximately) exponential distribution of transcript abundance this means that, in effect, very few genes dominate the analysis.
I started thinking about this issue with my student Nicolas Bray and we began by looking at the first obvious candidate for addressing the issue of domination by high variance genes: the Mahalanobis distance. Mahalanobis distance is an option in many heat map packages (e.g. in R), but has been used only rarely in publications (although there is some history of its use in the analyses of microarray data). Intuitively, Mahalanobis distance seeks to remedy the problem of genes with high variance among the samples dominating the distance calculation by appropriate normalization. This appears to have been the aim of the method in the Anders and Huber paper cited above, where the expression values are first normalized to obtain equal variance for each gene (the variance stabilization procedure). Mahalanobis distance goes a step further and better, by normalizing using the entire covariance matrix (as opposed to just its diagonal).
Intuitively, given a set of points in some dimension, the Mahalanobis distance is the Euclidean distance between the points after they have been transformed via a linear transformation that maps an ellipsoid fitted to the points to a sphere. Formally, I think it is best understood in the following slightly more general terms:
Given an 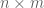
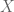
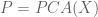




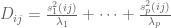
When 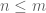


The Mahalanobis ellipses. In this figure the distance shown is from every point to the center (mean of the point) rather than between pairs of points. Mahalanobis distance is sometimes defined in this way. The figure is reproduced from this website. Note that the Anders-Huber heat map produces distances looking only at the variance in each direction (in this case horizontal and vertical) which assumes that the gene expression values are independent, or equivalently that the ellipse is not rotated.
It is interesting to note that D is defined even when 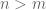
The cutoff p involves ignoring the last few principal components. The reason one might want to do this is that the last few principal components presumably correspond to noise in the data. Amplifying this noise and treating it the same as the signal is not desirable. This is because as p increases the denominators 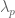
We’re still not completely happy with Mahalanobis distance. For example, unlike the Jensen-Shannon metric, it does not provide a metric over probability distributions. In functional genomics, almost all *Seq assays produce an output which is a (discrete) probability distribution (for example in RNA-Seq the output after quantification is a probability distribution on the set of transcripts). So making heat maps for such data seems to not be entirely trivial…
Does any of this matter?
The landmark Michael Eisen et al. paper “Cluster analysis and the display of genome-wide expression patterns“, PNAS 95 (1998), 14863–14868 describing the “Eisengram” was based on correlation as the distance measure between expression vectors. This has a similar problem to the issues we discussed above, namely that abundant genes are weighted more heavily in the distance measure, and therefore they define the characteristics of the heat map. Yet the Eisengram and its variants have proven to be extremely popular and useful. It is fair to ask whether any of the issues I’ve raised matter in practice.
Depends. In many papers the heat map is a visualization tool intended for a qualitative exploration of the data. The issues discussed here touch on quantitative aspects, and in some applications changing distance measures may not change the qualitative results. Its difficult to say without reanalyzing data sets and (re)creating the heat maps with different parameters. Regardless, as expression technology continues to transition from microarrays to RNA-Seq, the demand for quantitative results is increasing. So I think it does matters how heat maps are made. Of course its easy to ridicule Handan Wand for her quilt plots, but I think those guilty of pasting ad-hoc heat maps based on arbitrary distance measures in their papers are really the ones that deserve a public spanking.
P.S. If you’re going to make your own heat map, after adhering to sound statistics, please use a colorblind-friendly palette.
P.P.S. In this post I have ignored the issue of clustering, namely how to order the rows and columns of heat maps so that similar expression profiles cluster together. This goes along with the problem of constructing meaningful dendograms, a visualization that has been a major factor in the popularization of the Eisengram. The choice of clustering algorithm is just as important as the choice of similarity measure, but I leave this for a future post.

14 comments
Comments feed for this article
January 19, 2014 at 5:35 pm
Damian Kao
Great post. I think the decision to consider your normalized transcript abundance as absolute or relative values is a important one. If you consider them as relative values, then one can simply create a heatmap from standardized values (ie. number of st. deviations from mean).
And the decision to consider them absolute or relative stems from the multi-mapping reads strategy used. If only uniquely mapped reads were used, then there really is no point to consider them as absolute values as the resulting abundances will not be representative of the real biological abundance (transcripts with common sequence regions will be under-counted).
January 19, 2014 at 9:10 pm
Jason Merkin
Nice post, Lior. I have two things I would like to mention. First, since you mention my paper, I would like to add that when we did the clustering analysis in the Merkin et al paper, we tried a number of different distance metrics (though not mahalanobis–I will try it) that all gave similar results but opted to show jsd because of some of the information theoretic interpretations.
Second (and more of a question), you discuss choice of distance metric as it pertains to gene expression, which has a large dynamic range from fractions of transcripts per million to hundreds or thousands. What about splicing, where if you are analyzing isoform ratios, the values are bounded by [0, 1]? While I can come up with some contrived cases with very few events that have low inclusion ratios and one or two that have high values, generally you are considering hundreds or thousands of exons with a variety of inclusion ratios so no one value will be that much larger and dominate like in gene expression.
January 19, 2014 at 9:20 pm
Lior Pachter
Good question about splicing inclusion ratios- I haven’t thought about that setting and I’m not sure anyone else has. The issue with gene expression is not so much the variability in magnitude of expression, but the fact that the variance scales (super) linearly with expression. So high expression means high variance, and there is a large difference in the variance observed for low vs. highly expressed genes. I suspect that this issue will also arise in splicing ratios: some exons may always be spliced out/in (variance 0) whereas others uniformly at random (variance 1/12). So while you are right that there is an upper bound in this case on the variance, I still think the relative difference might matter. But I’m not sure- it is an interesting question to look into.
Many thanks for your comment: I’m curious to find out whether Mahalanobis does indeed make a difference.
January 19, 2014 at 10:00 pm
Dario Strbenac
There’s also a model-based clustering algorithm for RNA-seq data :
http://bioinformatics.oxfordjournals.org/content/30/2/197.long
Although the documentation is minimal and some features used to make the figures in the journal article aren’t implemented. I’m also not sure there are serious problems with using existing algorithms for the data type to warrant a new one.
January 19, 2014 at 10:06 pm
David Robinson
You didn’t mention the most absurd element of the quilt plot article. They didn’t provide R code: they provided *screenshots* of their R code. So they thought it was less effort to retype all their functions than to type “clustering=FALSE, dendrogram=FALSE”
January 24, 2014 at 4:01 am
Friday Coffee Break « Nothing in Biology Makes Sense!
[…] plot” which is really just a simple heatmap… but it spurs Lior Pachter to ask just what measures should go into a heatmap comparing gene expression data sets. From […]
February 1, 2014 at 5:57 pm
David Lovell
Great post!
Quilting aside, I would argue strongly that Euclidean distance, Pearson’s correlation or Spearman’s (rank) correlation are not appropriate for data that carry only relative information… in fact, they are known (largely outside molecular bioscience) to fall prey to Spurious Correlation (https://en.wikipedia.org/wiki/Spurious_correlation)
I suggest that for this kind of compositional data *proportionality* is the appropriate measure of pairwise association between components:
https://www.researchgate.net/publication/251878910_Have_you_got_things_in_proportion_A_practical_strategy_for_exploring_association_in_high-dimensional_compositions?ev=prf_pub
…and Aitchison’s distance is the right metric to use between compositions.
Aitchison’s distance is related to (symmetric) Kullback-Liebler divergence (as an upper bound).
June 17, 2014 at 1:00 am
Dario Strbenac
If p = n, then the distance D is the Mahalanobis distance. When p < n, is there a different name for it ? Also, do the singular values correspond to P or X ?
July 28, 2014 at 9:22 am
Anonymous Postdoc
Hi Lior,
Thank you for an interesting post as always. I have a question about the statement “Mahalanobis distance seeks to remedy the problem of genes with high variance among the samples dominating the distance calculation by appropriate normalization”. Are you sure that “high variance genes dominating the distance calcualtion” is necessarily a bad thing? Intuitively, low variance genes often won’t contain a lot of information, because the useful signal will inevitabley be drowned out by measurement error?
As a side note, would it make sense to test the various distance metrics in real data? I.e. applying them to some existing high throughput datasets, like TCGA. CCLE or GTEx and see how well various distance metrics recapitulate the expected structure of the data (based on cancer subtype, tissue of origin or any similar condition that you would expect a “good” distance metric to recapitulate).
Thanks again for posting and forgiveness if I have misunderstood your point.
August 3, 2014 at 7:00 pm
Dario Strbenac
Think about three clusters for four times. One has mean 100, 150, 200, 50. The second has mean 5, 10, 10, 5 and the third has mean 10, 5, 2, 1. Most genes which originate from the first cluster will have a much larger distance between them than genes from the other two clusters. This causes the two low expression clusters to be always clustered together, even though they have distinct biological causes. The cluster with large expression will be fragmented into many clusters, although it should be a single cluster. I found that transforming the data using a regularised logarithm transformation and using existing algorithms for microarrays gives adequate clustering in this scenario.
April 7, 2015 at 1:57 pm
Sander W. van der Laan
Hi,
Nice post. I have been surfing the internet to find an answer: what would be the most appropriate way to cluster and later visualise 380+ patients with measures for 29 proteins (in pg/L)? And shouldn’t one correct for covariates while clustering, for instance for age and sex?
Since the data scales are sometimes not overlapping, but I do want to compare the data, I Box-Cox transformed (because there are biologically plausible low levels) and than normalised the data. Than I just want to cluster the most comparable proteins (in the rows) and the most comparable patients in the columns. Intuitively I was thinking “correlation”. And that turns out to be “right”. But now about the distance and linkage…there I’m wondering what best to use… Even after reading your post I don’t know…
And another thing, I never see this done in papers: the correction for potential confounders prior to clustering. Is there a reason why?
Thanks!
Sander
April 7, 2015 at 2:01 pm
Sander W. van der Laan
Or rather…perhaps correlation is not such a good idea due to spurious correlations…
May 1, 2018 at 6:21 am
Tutorial: Brainmaps – Shibui Graphics
[…] on different clustering algorithms and methods used to calculate distances between samples. If used wisely, heatmaps can be a tool for discovery. Nevertheless, if we are only interested in simple […]
March 8, 2022 at 5:39 am
Mae Woods
Hi I only saw this post yesterday, but it’s still helpful.
Does anyone know if the heatmap has been extended to include hierarchical barcoded data. For example, RNAseq of single cells marked with labels n x A & m x B where n,m are the number of unique labels, the value of the label is binary indicating presence or absence, n<m & a set of cells that have been labeled with one value in A might not share the same label in B but not vice versa. Sorry if this is a stupid question, but can you add the root JSD (one for each variable) in this case? Or, is there an extension to the Mahalanobis distance with a label-aware compressive classification method? I saw a breaking paper yesterday & the closest thing to a heatmap was I think a plot of differential genes (possibly after imputation) identified by Seurat’s find cluster markers.
Thanks
This is such a good post, the heatmap in Merkin et al is so nice.