
Nature Publishing Group claims on its website that it is committed to publishing “original research” that is “of the highest quality and impact”. But when exactly is research “original”? This is a question with a complicated answer. A recent blog post by senior editor Dorothy Clyde at Nature Protocols provides insight into the difficulties Nature faces in detecting plagiarism, and identifies the issue of self plagiarism as particularly problematic. The journal tries to avoid publishing the work of authors who have previously published the same work or a minor variant thereof. I imagine this is partly in the interests of fairness, a service to the scientific community to ensure that researchers don’t have to sift through numerous variants of a single research project in the literature, and a personal interest of the journal in its aim to publish only the highest level of scholarship.
On the other hand, there is also a rationale for individual researchers to revisit their own previously published work. Sometimes results can be recast in a way that makes them accessible to different communities, and rethinking of ideas frequently leads to a better understanding, and therefore a better exposition. The mathematician Gian-Carlo Rota made the case for enlightened self-plagiarism in one of his ten lessons he wished he had been taught when he was younger:
3. Publish the same result several times
After getting my degree, I worked for a few years in functional analysis. I bought a copy of Frederick Riesz’ Collected Papers as soon as the big thick heavy oversize volume was published. However, as I began to leaf through, I could not help but notice that the pages were extra thick, almost like cardboard. Strangely, each of Riesz’ publications had been reset in exceptionally large type. I was fond of Riesz’ papers, which were invariably beautifully written and gave the reader a feeling of definitiveness.
As I looked through his Collected Papers however, another picture emerged. The editors had gone out of their way to publish every little scrap Riesz had ever published. It was clear that Riesz’ publications were few. What is more surprising is that the papers had been published several times. Riesz would publish the first rough version of an idea in some obscure Hungarian journal. A few years later, he would send a series of notes to the French Academy’s Comptes Rendus in which the same material was further elaborated. A few more years would pass, and he would publish the definitive paper, either in French or in English. Adam Koranyi, who took courses with Frederick Riesz, told me that Riesz would lecture on the same subject year after year, while meditating on the definitive version to be written. No wonder the final version was perfect.
Riesz’ example is worth following. The mathematical community is split into small groups, each one with its own customs, notation and terminology. It may soon be indispensable to present the same result in several versions, each one accessible to a specific group; the price one might have to pay otherwise is to have our work rediscovered by someone who uses a different language and notation, and who will rightly claim it as his own.
The question is: where does one draw the line?
I was recently forced to confront this question when reading an interesting paper about a statistical approach to utilizing controls in large-scale genomics experiments:
J.A. Gagnon-Bartsch and T.P. Speed, Using control genes to corrected for unwanted variation in microarray data, Biostatistics, 2012.
A cornerstone in the logic and methodology of biology is the notion of a “control”. For example, when testing the result of a drug on patients, a subset of individuals will be given a placebo. This is done to literally control for effects that might be measured in patients taking the drug, but that are not inherent to the drug itself. By examining patients on the placebo, it is possible to essentially cancel out uninteresting effects that are not specific to the drug. In modern genomics experiments that involve thousands, or even hundreds of thousands of measurements, there is a biological question of how to design suitable controls, and a statistical question of how to exploit large numbers of controls to “normalize” (i.e. remove unwanted variation) from the high-dimensional measurements.
Formally, one framework for thinking about this is a linear model for gene expression. Using the notation of Gagnon-Bartsch & Speed, we have an expression matrix 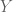
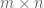
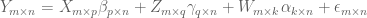
Here X is a matrix describing various conditions (also called factors) and associated to it is the parameter matrix 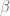

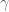
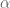
Linear models have been applied to gene expression analysis for a very long time; I can think of papers going back 15 years. But They became central to all analysis about a decade ago, specifically popularized with the Limma package for microarray data analysis. In an important paper in 2007, Leek and Storey focused explicitly on the identification of hidden factors and estimation of their influence, using a method called SVA (Surrogate Variable Analysis). Mathematically, they described a procedure for estimating k and W and the parameters
Gagnon-Bartsch and Speed refine this idea by suggesting that it is better to infer W from controls. For example, house-keeping genes that are unlikely to correlate with the conditions being tested, can be used to first estimate W, and then subsequently all the parameters of the model can be estimated by linear regression. They term this two-step process RUV-2 (acronym for Remote Unwanted Variation) where the “2” designates that the procedure is a two-step procedure. As with SVA, the key to inferring W from the controls is to perform singular value decomposition (or more generally factor analysis). This is actually clear from the probabilistic interpretation of PCA and the observation that what it means to be a in the set of “control genes” C in a setting where there are no observed factors Z, is that
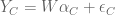
That is, for such control genes the corresponding
There is a tech report and also a preprint that follow up on the Gagnon-Bartsch & Speed paper; the tech report extends RUV-2 to a four step method RUV-4 (it also provides a very clear exposition of the statistics), and separately the preprint describes an extension to RUV-2 for the case where the factor of interest is also unknown. Both of these papers build on the original paper in significant ways and are important work, that to return to the original question in the post, certainly are on the right side of “the line”
The wrong side of the line?
The development of RUV-2 and SVA occurred in the context of microarrays, and it is natural to ask whether the details are really different for RNA-Seq (spoiler: they aren’t). In a book chapter published earlier this year:
D. Risso, J. Ngai, T.P. Speed, S. Dudoit, The role of spike-in standards in the normalization of RNA-Seq, in Statistical Analysis of Next Generation Sequencing Data (2014), 169-190.
the authors replace “log expression levels” from microarrays with “log counts” from RNA-Seq and the linear regression performed with Limma for RUV-2 with a Poisson regression (this involves one different R command). They call the new method RUV, which is the same as the previously published RUV, a naming convention that makes sense since the paper has no new method. In fact, the mathematical formulas describing the method are identical (and even in almost identical notation!) with the exception that the book chapter ignores Z altogether, and replaces
To be fair, there is one added highlight in the book chapter, namely the observation that spike-ins can be used in lieu of housekeeping (or other control) genes. The method is unchanged, of course. It is just that the spike-ins are used to estimate W. Although spike-ins were not mentioned in the original Gagnon-Bartsch paper, there is no reason not to use them with arrays as well; they are standard with Affymetrix arrays.
My one critique of the chapter is that it doesn’t make sense to me that counts are used in the procedure. I think it would be better to use abundance estimates, and in fact I believe that Jeff Leek has already investigated the possibility in a preprint that appears to be an update to his original SVA work. That issue aside, the book chapter does provide concrete evidence using a Zebrafish experiment that RUV-2 is relevant and works for RNA-Seq data.
The story should end here (and this blog post would not have been written if it had) but two weeks ago, among five RNA-Seq papers published in Nature Biotechnology (I have yet to read the others), I found the following publication:
D. Risso, J. Ngai, T.P. Speed, S. Dudoit, Normalization of RNA-Seq data using factor analysis of control genes or samples, Nature Biotechnology 32 (2014), 896-902.
This paper has the same authors as the book chapter (with the exception that Sandrine Dudoit is now a co-corresponding author with Davide Risso, who was the sole corresponding author on the first publication), and, it turns out, it is basically the same paper… in fact in many parts it is the identical paper. It looks like the Nature Biotechnology paper is an edited and polished version of the book chapter, with a handful of additional figures (based on the same data) and better graphics. I thought that Nature journals publish original and reproducible research papers. I guess I didn’t realize that for some people “reproducible” means “reproduce your own previous research and republish it”.
At this point, before drawing attention to some comparisons between the papers, I’d like to point out that the book chapter was refereed. This is clear from the fact that it is described as such in both corresponding authors’ CVs.
How similar are the two papers?
Final paragraph of paper in the book:
Internal and external controls are essential for the analysis of high-throughput data and spike-in sequences have the potential to help researchers better adjust for unwanted technical effects. With the advent of single-cell sequencing [35], the role of spike-in standards should become even more important, both to account for technical variability [6] and to allow the move from relative to absolute RNA expression quantification. It is therefore essential to ensure that spike-in standards behave as expected and to develop a set of controls that are stable enough across replicate libraries and robust to both differences in library composition and library preparation protocols.
Final paragraph of paper in Nature Biotechnology:
Internal and external controls are essential for the analysis of high-throughput data and spike-in sequences have the potential to help researchers better adjust for unwanted technical factors. With the advent of single-cell sequencing27, the role of spike-in standards should become even more important, both to account for technical variability28 and to allow the move from relative to absolute RNA expression quantification. It is therefore essential to ensure that spike- in standards behave as expected and to develop a set of controls that are stable enough across replicate libraries and robust to both differences in library composition and library preparation protocols.
Abstract of paper in the book:
Normalization of RNA-seq data is essential to ensure accurate inference of expression levels, by adjusting for sequencing depth and other more complex nuisance effects, both within and between samples. Recently, the External RNA Control Consortium (ERCC) developed a set of 92 synthetic spike-in standards that are commercially available and relatively easy to add to a typical library preparation. In this chapter, we compare the performance of several state-of-the-art normalization methods, including adaptations that directly use spike-in sequences as controls. We show that although the ERCC spike-ins could in principle be valuable for assessing accuracy in RNA-seq experiments, their read counts are not stable enough to be used for normalization purposes. We propose a novel approach to normalization that can successfully make use of control sequences to remove unwanted effects and lead to accurate estimation of expression fold-changes and tests of differential expression.
Abstract of paper in Nature Biotechnology:
Normalization of RNA-sequencing (RNA-seq) data has proven essential to ensure accurate inference of expression levels. Here, we show that usual normalization approaches mostly account for sequencing depth and fail to correct for library preparation and other more complex unwanted technical effects. We evaluate the performance of the External RNA Control Consortium (ERCC) spike-in controls and investigate the possibility of using them directly for normalization. We show that the spike-ins are not reliable enough to be used in standard global-scaling or regression-based normalization procedures. We propose a normalization strategy, called remove unwanted variation (RUV), that adjusts for nuisance technical effects by performing factor analysis on suitable sets of control genes (e.g., ERCC spike-ins) or samples (e.g., replicate libraries). Our approach leads to more accurate estimates of expression fold-changes and tests of differential expression compared to state-of-the-art normalization methods. In particular, RUV promises to be valuable for large collaborative projects involving multiple laboratories, technicians, and/or sequencing platforms.
Abstract of Gagnon-Bartsch & Speed paper that already took credit for a “new” method called RUV:
Microarray expression studies suffer from the problem of batch effects and other unwanted variation. Many methods have been proposed to adjust microarray data to mitigate the problems of unwanted variation. Several of these methods rely on factor analysis to infer the unwanted variation from the data. A central problem with this approach is the difficulty in discerning the unwanted variation from the biological variation that is of interest to the researcher. We present a new method, intended for use in differential expression studies, that attempts to overcome this problem by restricting the factor analysis to negative control genes. Negative control genes are genes known a priori not to be differentially expressed with respect to the biological factor of interest. Variation in the expression levels of these genes can therefore be assumed to be unwanted variation. We name this method “Remove Unwanted Variation, 2-step” (RUV-2). We discuss various techniques for assessing the performance of an adjustment method and compare the performance of RUV-2 with that of other commonly used adjustment methods such as Combat and Surrogate Variable Analysis (SVA). We present several example studies, each concerning genes differentially expressed with respect to gender in the brain and find that RUV-2 performs as well or better than other methods. Finally, we discuss the possibility of adapting RUV-2 for use in studies not concerned with differential expression and conclude that there may be promise but substantial challenges remain.
Many figures are also the same (except one that appears to have been fixed in the Nature Biotechnology paper– I leave the discovery of the figure as an exercise to the reader). Here is Figure 9.2 in the book:

The two panels appears as (b) and (c) in Figure 4 in the Nature Biotechnology paper (albeit transformed via a 90 degree rotation and reflection from the dihedral group):

Basically the whole of the book chapter and the Nature Biotechnology paper are essentially the same, down to the math notation, which even two papers removed is just a rehashing of the RUV method of Gagnon-Bartsch & Speed. A complete diff of the papers is beyond the scope of this blog post and technically not trivial to perform, but examination by eye reveals one to be a draft of the other.
Although it is acceptable in the academic community to draw on material from published research articles for expository book chapters (with permission), and conversely to publish preprints, including conference proceedings, in journals, this case is different. (a) the book chapter was refereed, exactly like a journal publication (b) the material in the chapter is not expository; it is research, (c) it was published before the Nature Biotechnology article, and presumably prepared long before, (d) the book chapter cites the Nature Biotechnology article but not vice versa and (e) the book chapter is not a particularly innovative piece of work to begin with. The method it describes and claims to be “novel”, namely RUV, was already published by Gagnon-Bartsch & Speed.
Below is a musical rendition of what has happened here:

10 comments
Comments feed for this article
September 12, 2014 at 2:07 am
John Pakala
This is what happens when the research is evaluated base on _number_ of papers/articles/books/chapters published.
June 4, 2015 at 9:19 am
cbouyio
… and we are just in the beginning…
September 12, 2014 at 7:19 am
Chirag Nepal
Nice one ! Interesting to see how Nat Biotechnology would respond to this. I seriously hope Nat Biotechnology will give their opinion on it. Probably put all the blame on authors.
September 12, 2014 at 8:01 am
Jeff Leek
Lior
Thanks for saying nice things about SVA. I’m out of the office, but I wanted to quickly clarify the timeline of events. Risso et al posted their RUVseq
R package several months ago and I saw the R package on Bioconductor. When I noticed it was related to my work on SVA I communicated with them and they sent me a preprint of their paper. I then wrote my svaseq paper
in response and posted the preprint (which was just accepted at Nucleic Acids Research). But their paper was accepted before my preprint appeared.
Cheers,
Jeff Leek
September 12, 2014 at 8:04 am
Lior Pachter
Thanks for clarifying Jeff. That is roughly what I had assumed when I ran across your preprint. Congratulations on the acceptance to NAR!
September 12, 2014 at 8:07 am
homolog.us
‘Self-plagiarism’ is an oxymoron. The real problem is in bastardization of the word ‘plagiarism’ to mean ‘copyright violation’, which is entirely different from original meaning of the word –
http://www.homolog.us/blogs/students/2013/04/26/history-of-plagiarism/
Nature cares about ‘self’ copyright violation. Scholars should make sure the word reverts to its original meaning instead of the meaning that makes the lives of commercial journals better.
September 15, 2014 at 5:55 am
John Pakala
It’s time to criminalise serious scientific misconduct!
========================================
Why should research misconduct be illegal?
After 30 years of observing how science deals with the problem, I have sadly come to the conclusion that it should be a crime, for three main reasons. First, in a lot of cases, people have been given substantial grants to do honest research, so it really is no different from financial fraud or theft. Second, we have a whole criminal justice system that is in the business of gathering and weighing evidence – which universities and other employers of researchers are not very good at. And finally, science itself has failed to deal adequately with research misconduct.
(from: http://www.newscientist.com/article/mg22329864.100-its-time-to-criminalise-serious-scientific-misconduct.html#.VBbhCXWSy90 )
========================================
June 4, 2015 at 9:29 am
cbouyio
We pay dearly for this inadequacy to deal with scientific contact in two ways. Once, obviously, by generating obstacles in terms of scientific progress and generation of knew knowledge (who should really care about that, everybody should only look their career and then the “invisible hand” of “self regulation” will fix everything)
Secondly, in an indirect way with the increasing mistrust of the general public to scientific endeavours and the rising movements of science deniers (creationist, vaccine deniers etc.).
September 16, 2014 at 3:51 am
John Pakala
Guidelines for “text recycling”:
Click to access BioMed%20Central_text_recycling_editorial_guidelines.pdf
November 25, 2015 at 8:03 pm
tangboyun
The scatter matrix plot for PCA in the RUVseq paper is somewhat problematic. The author directly performed the PCA on the p >> n matrix instead of transpose it first.