
“An entertaining freshness… Tic Tac!” This is Ferrero‘s tag line for its most successful product, the ubiquitous Tic Tac. And the line has stuck. As WikiHow points out in how to make your breath fresh: first buy some mints, then brush your teeth.
One of the amazing things about Tic Tacs is that they are sugar free. Well… almost not. As the label explains, a single serving (one single Tic Tac) contains 0g of sugar (to be precise, less than 0.5g, as explained in a footnote). In what could initially be assumed to be a mere coincidence, the weight of a single serving is 0.49g. It did not escape my attention that 0.50-0.49=0.01. Why?

To understand it helps to look at the labeling rules of the FDA. I’ve reproduced the relevant section (Title 21) below, and boldfaced the relevant parts:
TITLE 21–FOOD AND DRUGS
CHAPTER I–FOOD AND DRUG ADMINISTRATION
DEPARTMENT OF HEALTH AND HUMAN SERVICES
SUBCHAPTER B–FOOD FOR HUMAN CONSUMPTION (c) Sugar content claims –(1) Use of terms such as “sugar free,” “free of sugar,” “no sugar,” “zero sugar,” “without sugar,” “sugarless,” “trivial source of sugar,” “negligible source of sugar,” or “dietarily insignificant source of sugar.” Consumers may reasonably be expected to regard terms that represent that the food contains no sugars or sweeteners e.g., “sugar free,” or “no sugar,” as indicating a product which is low in calories or significantly reduced in calories. Consequently, except as provided in paragraph (c)(2) of this section, a food may not be labeled with such terms unless:
(i) The food contains less than 0.5 g of sugars, as defined in 101.9(c)(6)(ii), per reference amount customarily consumed and per labeled serving or, in the case of a meal product or main dish product, less than 0.5 g of sugars per labeled serving; and
(ii) The food contains no ingredient that is a sugar or that is generally understood by consumers to contain sugars unless the listing of the ingredient in the ingredient statement is followed by an asterisk that refers to the statement below the list of ingredients, which states “adds a trivial amount of sugar,” “adds a negligible amount of sugar,” or “adds a dietarily insignificant amount of sugar;” and
(iii)(A) It is labeled “low calorie” or “reduced calorie” or bears a relative claim of special dietary usefulness labeled in compliance with paragraphs (b)(2), (b)(3), (b)(4), or (b)(5) of this section, or, if a dietary supplement, it meets the definition in paragraph (b)(2) of this section for “low calorie” but is prohibited by 101.13(b)(5) and 101.60(a)(4) from bearing the claim; or
(B) Such term is immediately accompanied, each time it is used, by either the statement “not a reduced calorie food,” “not a low calorie food,” or “not for weight control.”
It turns out that Tic Tacs are in fact almost pure sugar. Its easy to figure this out by looking at the number of calories per serving (1.9) and multiplying the number of calories per gram of sugar (3.8) by 0.49 => 1.862 calories. 98% sugar! Ferrero basically admits this in their FAQ. Acting completely within the bounds of the law, they have simply exploited an arbitrary threshold of the FDA. Arbitrary thresholds are always problematic; not only can they have unintended consequences, but they can be manipulated to engineer desired outcomes. In computational biology they have become ubiquitous, frequently being described as “filters” or “pre-processing steps”. Regardless of how they are justified, thresholds are thresholds are thresholds. They can sometimes be beneficial, but they are dangerous when wielded indiscriminately.
There is one type of thresholding/filtering in used in RNA-Seq that my postdoc Bo Li and I have been thinking about a bit this year. It consists of removing duplicate reads, i.e. reads that map to the same position in a transcriptome. The motivation behind such filtering is to reduce or eliminate amplification bias, and it is based on the intuition that it is unlikely that lightning strikes the same spot multiple times. That is, it is improbable that many reads would map to the exact same location assuming a model for sequencing that posits selecting fragments from transcripts uniformly. The idea is also called de-duplication or digital normalization.
Digital normalization is obviously problematic for high abundance transcripts. Consider, for example, a transcripts that is so abundant that it is extremely likely that at least one read will start at every site (ignoring the ends, which for the purposes of the thought experiment are not relevant). This would also be the case if the transcript was twice as abundant, and so digital normalization would prevent the possibility for estimating the difference. This issue was noted in a paper published earlier this year by Zhou et al. The authors investigate in some detail the implications of this problem, and quantify the bias it introduces in a number of data sets. But a key question not answered in the paper is what does digital normalization actually do?
To answer the question, it is helpful to consider how one might estimate the abundance of a transcript after digital normalization. One naive approach is to just count the number of reads after de-duplication, followed by normalization for the length of the transcript and the number of reads sequenced. Specifically if there are n sites where a read might start, and k of the sites had at least one read, then the naive approach would be to use the estimate 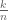
Let X be a random variable that denotes the number of sites on a transcript of length n that are covered in a random sequencing experiment, where the number of reads starting at each site of the transcript is Poisson distributed with parameter c (i.e., the average coverage of the transcript is c). Note that

The maximum likelihood estimate for c can also be obtained by the method of moments, which is to set

from which it is easy to see that
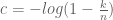
This is the same as the (derivation of the) Jukes-Cantor correction in phylogenetics where the method of moments equation is replaced by 

The point here, as noticed by Bo Li, is that since 


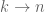

Digital normalization as proposed by Brown et al. involves possibly thresholding at more than one read per site (for example choosing a threshold C and removing all but at most C reads at every site). But even this modified heuristic fails to adequately relate to a probabilistic model of sequencing. One interesting and easy exercise is to consider the second or higher order Taylor approximations. But a more interesting approach to dealing with amplification bias is to avoid thresholding per se, and to instead identify outliers among duplicate reads and to adjust them according to an estimated distribution of coverage. This is the approach of Hashimoto et al. in a the paper “Universal count correction for high-throughput sequencing” published in March in PLoS One. There are undoubtedly other approaches as well, and in my opinion the issue will received renewed attention in the coming year as the removal of amplification biases in single-cell transcriptome experiments becomes a priority.
As mentioned above, digital normalization/de-duplication is just one of many thresholds applied in a typical RNA-Seq “pipeline”. To get a sense of the extent of thresholding, one need only scan the (supplementary?) methods section of any genomics paper. For example, the GEUVADIS RNA-Seq consortium describe their analysis pipeline as follows:
“We employed the JIP pipeline (T.G. & M.S., data not shown) to map mRNA-seq reads and to quantify mRNA transcripts. For alignment to the human reference genome sequence (GRCh37, autosomes + X + Y + M), we used the GEM mapping suite24 (v1.349 which corresponds to publicly available pre-release 2) to first map (max. mismatches = 4%, max. edit distance = 20%, min. decoded strata = 2 and strata after best = 1) and subsequently to split-map (max.mismatches = 4%, Gencode v12 and de novo junctions) all reads that did not map entirely. Both mapping steps are repeated for reads trimmed 20 nucleotides from their 3′-end, and then for reads trimmed 5 nucleotides from their 5′-end in addition to earlier 3′-trimming—each time considering exclusively reads that have not been mapped in earlier iterations. Finally, all read mappings were assessed with respect to the mate pair information: valid mapping pairs are formed up to a maximum insert size of 100,000 bp, extension trigger = 0.999 and minimum decoded strata = 1. The mapping pipeline and settings are described below and can also be found in https://github.com/gemtools, where the code as well as an example pipeline are hosted.”
This is not a bad pipeline- the paper shows it was carefully evaluated– and it may have been a practical approach to dealing with the large amount of RNA-Seq data in the project. But even the first and seemingly innocuous thresholding to trim low quality bases from the ends of reads is controversial and potentially problematic. In a careful analysis published earlier this year, Matthew MacManes looked carefully at the effect of trimming in RNA-Seq, and concluded that aggressive trimming of bases below Q20, a standard that is frequently employed in pipelines, is problematic. I think his Figure 3, which I’ve reproduced below, is very convincing:

It certainly appears that some mild trimming can be beneficial, but a threshold that is optimal (and more importantly not detrimental) depends on the specifics of the dataset and is difficult or impossible to determine a priori. MacManes’ view (for more see his blog post on the topic) is consistent with another paper by Del Fabbro et al. that while seemingly positive about trimming in the abstract, actually concludes that “In the specific case of RNA-Seq, the tradeoff between sensitivity (number of aligned reads) and specificity (number of correctly aligned reads) seems to be always detrimental when trimming the datasets (Figure S2); in such a case, the modern aligners, like Tophat, seem to be able to overcome low quality issues, therefore making trimming unnecessary.”
Alas, Tic Tac thresholds are everywhere. My advice is: brush your teeth first.

20 comments
Comments feed for this article
September 11, 2014 at 4:12 am
Titus Brown
Hi Lior,
I read your blog post first with interest and then with dismay – I
feel like there is a tremendous misunderstanding at the heart of your
description of digital normalization. In fact, your blog post has
nothing to do with digital normalization and completely misrepresents
it.
First, digital normalization isn’t deduplication, and I agree with all
your comments about deduplication.
Second, the description of digital normalization (the “naive
approach” is simply wrong; moreover, digital normalization makes no
assumptions about where a read might start, and does not take into
account the total number of reads in any way.
Third, no assumption about the distribution of reads from transcripts
is made in any way by digital normalization.
Fourth, digital normalization isn’t intended to relate to a
probabilistic model of sequencing. It’s solely concerned with
observing the saturation of assembly graphs.
Finally, I certainly wouldn’t recommend doing differential
expression analysis after digital normalization; in fact, I don’t
think anyone does. It would be pointless! Diginorm and its
derivatives and compadres are primarily intended for the purpose of
achieving an assembly in low memory; several studies have shown that
it does that very well, which is one reason why Trinity has adopted a
similar approach.
Happy to discuss further. There are many, many problems with digital
normalization, and I’m always looking to learn more!
–titus
September 11, 2014 at 4:26 am
Lior Pachter
Titus,
I appreciate that you responded on the blog, and I’m glad that we agree about de-duplication. My understanding of digital normalization was based on your blog post about it and on the arXiv paper I reference in my post. The way you explain it, I believe it is exactly de-duplication, albeit more general, because de-duplication refers to the removal of all duplicates, whereas digital normalization removes all but C, and none if there are less than C duplicates of a read (page 3 of your arXiv preprint).
I understand that one of the primary applications of removing duplicate reads is assembly, and indeed it may indeed have been used successfully in that regard. But your blog post refers specifically to abundance estimation, and indeed it was my understanding that this was also an intended application, especially in regards to metagenomics (but also RNA-Seq). I also have to add that I don’t see any problem with using de-duplication or digital normalization for RNA-Seq– it may be beneficial in quantifying robustly with respect to amplification bias, especially for low abundance transcripts, and as you point out it has other advantages (e.g. memory). My blog post merely explains what it means statistically to do it, and in doing so explains how one might improve on the method. Specifically, rather than discarding reads and proceeding with a pipeline, its better to think in another way, namely in transcriptome space in terms of the number of sites that have at least one read mapping to them. In no way does the math imply anything about digital normalization requiring the total number of reads, and the issue about whether we are talking about removing duplicates before or after mapping does not change the math either. So perhaps we are not exactly on the same page regarding either what you understand from my blog post, or what I understand from your descriptions of digital normalization.
Thanks again for your prompt reply here.
September 11, 2014 at 4:35 am
Titus Brown
Hi again Lior,
The abundance estimates in the blog post are calculated by mapping to
the known reference, and the median k-mer count estimates in the
preprint are used to demonstrate that there is a correlation between
that particular estimator and mapping. We then proceed to use them to
downsample the data for assembly — not for counting.
Nobody is (or should be, at least) using digitally normalized data to
do any abundance estimation anywhere. If you’ve managed to find
someone doing this, let me at ’em ;). Perhaps this is the central
point of misunderstanding?
(Isn’t it 4:30am in California!?)
–titus
September 11, 2014 at 5:14 am
Lior Pachter
First, I’m glad we are finally on the same page 🙂
If you’d like to call what I’m talking about in my blog-post “de-duplication” and reserve the term “digital normalization” for the same procedure but only as applied to assembly and not quantification, that’s certainly your prerogative- I will not debate you on your choice of semantics. I also acknowledge that your semantics is consistent with how you have been using digital normalization, or have intended it to be used.
However as I’ve already explained, my understand of the procedure was based on the text you yourself wrote, namely the arXiv preprint referred to in my post, and specifically page 3 that defines the term “digital normalization”:
You provide pseudocode that is
for read in dataset:
if estimated_coverage(read)<C:
accept(read)
else:
discard(read)
You go on to say "The net effect of this procedure, which we call digital normalization, is to normalize the coverage distribution of data sets." Nowhere in the section are you talking about assembly, although you do later give examples of how this procedure may improve assembly quality and feasibility.
I'm glad that this blog post has clarified your intent for digital normalization, and has hopefully educated readers about when de-duplication may be suitable. Although I do want to emphasize that quantification is a key part of assembly of transcriptomes, and therefore even for assembly, it would make (more) sense to quantify using a suitable model for data that has been de-duplicated (to use the preferred term now), and that is presumably not the case when assemblers that quantify by #reads are fed digitally normalized data. In other words, I think that even with your intent and definition of "digital normalization", the math of this post is relevant 🙂
September 11, 2014 at 6:32 am
Titus Brown
Hi Lior,
I generally think of de-duplication as referring to the removal of artificially duplicated reads (ADRs), which are an artifact of colony construction. Typically these reads start at the same position in the reference and, as they are technical artifacts of the sequencing prep, typically differ little in their sequence between them. I understood your blog post to be about that, and while I haven’t spent much time on your math, I of course trust you (although I’m happy to review it if you think it would aid in my understanding). These ADRs challenge abundance estimation, as noted in one of the original papers on them, Gomez-Alvarez et al (pmid 19587772). The process of finding these and removing them is confounded by high coverage and sequencing bias, as you note above.
The procedure outlined in the diginorm paper is about removing duplicate *information* from the read data set, not deduplication of ADRs. No attention is paid to the position of the read start, which is how ADR duplication typically works. Because diginorm explicitly eliminates all abundance information above a certain threshold, it would be an incredibly stringent way of doing de-duplication; it also would miss most of the ADRs that were low abundance. Bad all around, at both a theoretical and a practical level.
So I think there is a confusion in here about deduplication of reads vs removal of duplication information. The description of diginorm in your initial post is incorrect – see “more than C reads at every site” — and I infer that you have misunderstood the basic algorithm, as well as some of its implications.
With respect to assembly, you correctly note that abundance estimation is one of the things that assembler can take into account. We have been surprised (but in the end, not puzzled 🙂 to see that normalization procedures often end up improving assemblies in various ways; I think this is due to the dependence of many assembly algorithms on poorly understood heuristics.
best,
–titus
September 11, 2014 at 9:05 am
Lior Pachter
Hi Titus,
I think this exchange has been helpful in clarifying things, so I am continuing it. First, I do recommend you understand the math in my post; as a general principle its helpful to read and understand the posts in order to comment on them. I don’t think I’m confused about digital normalization. I understand that reads are removed in the procedure without first mapping them to a reference, by virtue of examining the k-mer frequencies inside them. I also understand that the goal is to remove reads in regions with high coverage, and not explicitly to remove duplicate reads. I get all of that. But its also true that digital normalization is essentially equivalent to de-duplication if you think of a k-mer as a “read” instead of the read object itself. Basically k-mers at high frequencies get deleted (via the process of removing reads) but the way in which it is technically achieved is not relevant for the math. Since these k-mers are what are used in assembly, and quantification is important for assembly (frequently achieved by smoothing k-mer counts across edges), I continue to insist on the relevance of the post. I do concede that I should have more clearly explained this point, and the rationale for equivalence between de-duplication and digital normalization in the original post. I didn’t think the semantics would bother people.
Indeed, taking a step back, I think I was affording a measure of respect to digital normalization by crediting it as a methodological idea rather than a heuristic. Specifically, I see it as the idea of improving robustness by discarding quantity of reads (or k-mers) via discretizing to the presence or absence of such objects. I think what has come across in your posts (and in Tracy’s) is that you think of digital normalization more as a “procedure” (first discard reads using the described approach for the purpose of assembly, then put them back in for abundance estimation). I honestly think my abstraction of it via identifying it with de-duplication is the right way to think about it. And I probably do disagree with you that it is not a good idea for quantification. Quite the contrary.. I believe it has much potential, especially for single-cell RNA-Seq. But one needs to think about how to use the information it provides.
One final point: there is no dichotomy between what has been called “artificial duplicated reads” and identical reads due to random sequencing; certainly not in RNA-Seq. The paper by Hashimoto et al. makes that clear.
September 11, 2014 at 4:40 am
Titus Brown
BTW, if you want to see how we use diginorm for mRNAseq, please see: https://khmer-protocols.readthedocs.org/en/latest/mrnaseq/index.html, esp #2 and #7.
September 11, 2014 at 6:36 am
tracykteal
What you’re actually discussing in this post is the filtering of artificial replicates (Identifying and Removing Artificial Replicates from 454 Pyrosequencing Data http://cshprotocols.cshlp.org/content/2010/4/pdb.prot5409.long and Systematic artifacts in metagenomes from complex microbial communities http://www.nature.com/ismej/journal/v3/n11/full/ismej200972a.html code: https://github.com/tracykteal/replicate-filter/). Artificial duplicates and replicates were discovered in 454 metagenomic data. As microbial communities are complex and depth of sampling low, the probability of reads sharing high percent similarity and starting at the same position is very, very low. These reads are instead *artificial* replicates that are generated during the emPCR step. To include these reads in an abundance analysis would be incorrect and the reads should therefore be removed before abundance calculations are made. CD-HIT has a version of this and MG-RAST includes the dereplication step in its pipeline. This is a well-acknowledged issue.
The point you mention about it being used in transcriptomics is absolutely true. As you mention the probabilities in a transcriptomic dataset are very different. In this case you expect many reads from the same region, so to use dereplication, especially with default parameters, would be inappropriate. It is likely still an issue in these datasets (at least for 454), but different parameters or potentially algorithms would need to be used to determine them. Your math demonstrates this well, and is a point we made in our papers and has been made in others.
Dereplication of artificial reads is designed to remove erroneous reads. I suppose that you could use digital normalization to remove artificial reads as well. It’s an interesting idea, but this is not its designed use case and is not how it is being used informatically or in the literature. The fundamental idea of digital normalization is that reads are not removed from abundance analysis, but they are removed at the computationally intensive steps of processes like metagenomic assembly and then mapped back and re-accounted for in abundance calculations.
September 11, 2014 at 10:14 am
homolog.us
Based on my understanding, Titus introduced ‘digital normalization’ to remove the erroneous reads so that assembly could proceed without saturating all available RAM. That used to be a big problem in the past, but later a number of other assembly programs took care of the same, making digital normalization unnecessary. I do not think the program was supposed to be used for counting and expression estimation.
However, Titus also marketed his program well and technicians-known-as-bioinformaticians, who do not want to learn any theory, use it as an absolutely necessary tool to solve every problem related to NGS.
September 11, 2014 at 10:16 am
homolog.us
Lior, nice transriptome assembly work with Sreeram and Tse BTW. I enjoyed the ISIT slides.
September 11, 2014 at 10:20 am
homolog.us
“Although I do want to emphasize that quantification is a key part of assembly of transcriptomes.”
As you understand, the transcriptome assembly problem is solved by people working on genome assembly as an afterthought, and they did not treat transcriptome assembly as a different (and difficult) problem. So, all those issues that you describe in ISIT slides had been completely overlooked, and researchers are very enamored with crap like Trinity.
September 11, 2014 at 11:11 am
Ariel Schwartz (@ArielSSchwartz)
Lior, is your transcriptome assembly software available for beta-testing?
I would like to try it out on some of our datasets.
September 11, 2014 at 11:24 am
Lior Pachter
Hi Ariel,
I’d first like to clarify that the algorithms and software were developed by Sreeram Kannan working together with David Tse and myself. Sreeram is about to start a position as assistant professor at the University of Washington this fall, and has been working hard to submit the paper. We are currently beta-testing ourselves, and I think it would be great if you could try it out. I will connect you with Sreeram by email. We expect to be able to distribute the software more widely in the near future.
September 12, 2014 at 1:43 am
Ariel Schwartz (@ArielSSchwartz)
Thanks. I’ll be looking forward to hearing from Sreeram.
Also, do you have a link to the aforementioned ISIT slides?
Regarding your post. I think the use of Digital Normalization in the title diverted the followup conversation away from the more important point you have made, which is that thresholds are everywhere, they are usually glanced over, but can have significant impact on the final analysis results. DN is just one minor example of that.
Even for RNA-Seq analysis, there are large number of thresholds that are being set at every step of the process, starting from sample QC, cluster generation (what threshold defines PF reads?), base calls, quality scores (now including Quality Score Binning with thresholds for the bins http://res.illumina.com/documents/products/whitepapers/whitepaper_datacompression.pdf), read trimming parameters, mapping score thresholds, and most importantly p-value thresholds for DE significance calls (some will also add a threshold on fold-change on top of that).
Some use of thresholds is probably unavoidable for practical purposes, but in my opinion algorithms should be designed with the goal of minimizing the number of thresholds that are need to be set, and with clear understanding of what effect these thresholds have on the final answer.
A somewhat philosophical question is why are thresholds so prevalent in general, and in computation biology in particular. I think part of the answer lies in our (especially biologists) expectation that an algorithm should provide a single correct answer to a complex experimental question given the input data provided (e.g. what is the correct linear sequence of a reference genome or transcriptome? which genes are up/down regulated in condition X vs Y?), when in reality the data cannot provide a single correct answer, or in many cases the question asked is inherently undefined (is there such thing as a single sequence that represents the correct and complete reference human genome?).
Computational Biology algorithms should strive not to provide a single correct answer based on a set of arbitrary thresholds, but rather a set of (partial) answers that can be explored by the users. The uncertainty in each step of a pipeline should be recorded as much as possible and passed on to the next processing step.
For example, instead of using hard base-call quality thresholds to trim sequences in fasta files, sequencer produce fastq files with quality scores, which record the uncertainty in the base calling procedure. Unfortunately, this is typically where the uncertainty information-exchange ends. Most assembly algorithms produce linear sequences as their output, even when a better answer will be represented by a graph or even a set of graphs, because that is what they are expected to do. Mapping algorithms (which mostly only work with linear references) are expected to designate exactly one alignment per read as Primary Alignment, even when mapping ambiguity exists. Even when mapping quality scores are provided in the BAM files, many expression estimation tools tend to ignore them as far as I know. Even though many expression estimation tools, can provide variance values with their FPKM/TPM/Count values, most DE tools ignore this information. In other words, instead of propagating the uncertainty throughout the analysis pipeline, every processing step relies on arbitrary thresholds to “simplify” the final answer and mask the ambiguity from the next step.
More research work should be dedicated to ways for representing and maintaining uncertainty information throughout the analysis pipelines by supporting graph-based representations of genomes, metagenomes and transcriptomes, multi-mapped reads, probabilistic read count estimates, etc.
Another big reason that thresholds are so popular is the fact that many algorithms are designed to work in batch mode. You are expected to provide all the data to an assembly algorithm, wait for a long time, and at the end of the process get your perfect linear sequence, which represents the one absolute underlying truth, which you will further try to annotate with the single correct set of transcript models, to which you will then assign absolute expression levels, etc.
What happens when new data becomes available after the analysis has completed (this is almost always the case now days, with the low cost of NGS)? Using the existing algorithms you more or less have to start all over again using all the data. This is also the case even without new data if you just want to change one of the thresholds in the pipeline.
Batch mode is many times also not scalable, with more data producing even worse results in some cases. This is where Digital Normalization comes into play. Instead of making the assembly algorithm more robust, the practical solution is to hide some of the (potentially redundant) information from it using some arbitrary coverage thresholds. A much more elegant approach would be to use an online assembly algorithm that keeps updating the assembly as more data is presented in a streaming fashion (from a MinION USB mybe?). Redundant data can be ignored when not providing useful information. Even when a truly online assembly is not feasible, assembly and other algorithms should be designed to support incremental improvement when new data is presented. In my work, I have avoided using DN for assembly of very large metagenomes using an incremental assembly approach instead. Some examples for online or incremental approaches that I have encountered include Updating RNA-Seq analyses after re-annotation (http://bioinformatics.oxfordjournals.org/content/early/2013/05/14/bioinformatics.btt197), Streaming fragment assignment for real-time analysis of sequencing experiments (http://www.nature.com/nmeth/journal/v10/n1/full/nmeth.2251.html), Incremental construction of FM-index for DNA sequences (http://bioinformatics.oxfordjournals.org/content/early/2014/08/26/bioinformatics.btu541.full). I am sure there are many more such examples, but they are still the exception and not the norm.
In my opinion, an ideal bioinformatics pipeline should strive to be able to continuously run end-to-end, while updating the analysis results as more data becomes available and as improvements to the algorithms are implemented. This is similar to the concept of continuous queries over data streams in the database world (http://dl.acm.org/citation.cfm?id=603884), but with the query being a biological question, and the data stream, sequence or other ‘omics data. This will entail a continuously updated graph-based representation of the genome, online transcript annotations, expression level estimations, and so on. This model is obviously a pipe-dream at this point, but on the other hand it can still serve as platform to strive-for when designing new algorithms, which in turn might help remove the need for some of the arbitrary thresholds currently in use.
September 12, 2014 at 8:08 am
Rob Patro
Hi Lior,
First, I’d like to congratulate Sreeram on his new job; this is great news! I’ve been very interested in your transcript assembly work since first hearing about it at GI last year. Would you be willing to share the slides referred to above? I’m looking forward to reading the paper and understanding your method. Thanks!
September 12, 2014 at 3:00 am
Titus Brown
Mostly agree. Note that, with digital normalization as implemented in khmer, you can always feed new data in while taking account of all of the old data — that is, it explicitly allows updating, for exactly the reasons you lay out. (Sorry to keep talking about diginorm, but it _is_ what we actually work on 🙂
September 12, 2014 at 3:01 am
Titus Brown
Lior, you might be interested in —
http://ged.msu.edu/downloads/2012-career-nsf-final.pdf (unfunded)
http://ged.msu.edu/downloads/2012-bigdata-nsf.pdf (funded)
–titus
September 22, 2014 at 8:53 pm
roye
A new preprint on this topic: http://biorxiv.org/content/early/2014/09/19/009407 (in the context of metagenomes)
September 25, 2014 at 10:49 am
homolog.us
Hello all, I posted the ISIT slides here –
http://www.homolog.us/blogs/blog/2014/09/09/tutorial-on-david-tses-work-on-dnarna-assembly-presented-at-isit-2014/
October 12, 2014 at 12:58 pm
Krishna
Geeze Lior, you scared me into thinking some people might be doing deduplication and then abundance calculations. That’s no way to spend a Sunday. I’m glad that people aren’t doing that… at least on purpose.