
In the August 2013 issue of Nature Biotechnology there were two back-to-back methods papers published in the area of network theory:
- Baruch Barzel & Albert-László Barabási, Network link prediction by global silencing of indirect correlations, Nature Biotechnology 31(8), 2013, p 720–725. doi:10.1038/nbt.2601.
- Soheil Feizi, Daniel Marbach, Muriel Médard & Manolis Kellis, Network deconvolution as a general method to distinguish direct dependencies in networks, Nature Biotechnology 31(8), 2013, p 726–733. doi:10.1038/nbt.2635.
This post is the first of a trilogy (part2, part3) in which my student Nicolas Bray and I tell the story of these papers and why we took the time to read them and critique them.
We start with the Barzel-Barabási paper that is about the applications of a model proposed by Barzel and his Ph.D. advisor, Ofer Biham (although all last names start with a B, Biham is not to be confused with Barabási):
In order to quantify connectivity in biological networks, Barzel and Biham proposed an experimental perturbation model in the paper Baruch Barzel & Ofer Biham, Quantifying the connectivity of a network: The network correlation function method, Phys. Rev. E 80, 046104 (2009) that forms the basis for network link prediction in Barzel-Barabási. In the context of biology, link prediction refers to the problem of identifying functional links between genes from data that may be confounded by indirect effects. For example, if gene A inhibits the expression of gene B, and also gene B inhibits the expression of gene C, then if the expression of A increases, it will decrease the expression of B, which in turn increase C. Therefore one might observe correlation in the expression levels of gene A and C, even though there is no direct interaction between them. The Barzel-Biham model is based on perturbation experiments. Assuming that a system of genes is in equilibrium, it is a model for the change in expression of one gene in response to a small perturbation in another.
The parameters in the Barzel-Biham model are entries in what they call a “local response matrix” S (any matrix with 
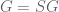
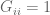
for a “global response matrix” G that can, in principle, be observed and used to infer the matrix S. The innovation of Barzel and Barabási is to provide an approximate formula for recovering S from G, specifically the formula

where D(M) denotes the operation setting off-diagonal elements of M to zero. A significant part of the paper is devoted to showing that the approximation (2) is good. Then they suggest that (2) can be used to infer direct causal links in regulatory networks from collections of expression experiments. Barzel and Barabási claim that the approximation formula (2) is necessary because exact inference of S from G requires solving the intractable system of 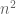
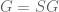
The assertion of intractability is based on the claim that the equations are coupled. They reason that since the naïve matrix inversion algorithm requires 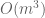
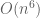
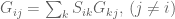
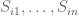
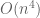
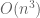
But there is more: while looking at the paper I had to take a quick bathroom break, and by the time I returned Nick had realized he could apply the Sherman-Morrison formula to obtain the following formula for the exact solution:
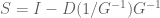
Here the operator “/” denotes element-wise division, a simple operation to execute, so that inferring S from G requires no more than inverting G and scaling it, a formula that is also much simpler and more efficient to compute than (2). [Added 2/23: Jordan Ellenberg pointed out the obvious fact that 
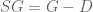

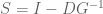


The approach to solving (4) has an implication that is even more important than the solution itself. It provides a dual formula for computing G from S according to (1), i.e. to simulate from the model. Using the same ideas as above, one finds that

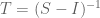
Unlike Barzel & Barabási that resorted to simulating with Michaelis-Menten dynamics in their study of performance of their approximation, using (4) we can efficiently simulate data directly from the model. One issue with Michaelis-Menten dynamics is that they make more sense for enzymatic networks as opposed to regulatory networks (for more on this see Karlebach, G. & Shamir, R. Modelling and analysis of gene regulatory networks, Nat Rev Mol Cell Biol 9, 770–780 (2008)), but in any case performance on such dynamics is hardly a validation of (2) since its mixing apples and oranges. So what happens when one simulates data from the Barzel-Biham model and then tries to recover the parameters?

A comparison of the standard method of regularized partial correlations with exact inference for the Barzel-Biham model. Random sparse graphs were generated according to the Erdös-Renyi graph model G(5000,p) where p was varied to assess performance at different graph densities (shown on x-axis) The y-axis shows the average AUROC obtained from 75 random trials at each density.
When examining simulations from the Barzel-Biham model with graphs on 5,000 nodes (see Figure above), we were surprised to discover that when adding even small amounts of noise, the exact algorithm (4) failed to recover the local response matrix from G (we also analyzed the approximation (3) and observed that it always resulted in performance inferior to (4), and that 5% of the time the correlation with the exact solution was negative). This sensitivity to noise is due to the term 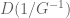

Some intuition for the behavior of 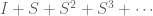
However, the difficulties with noise for the Barzel-Biham model go much deeper. While a constant signal-to-noise ratio, as assumed by Barzel and Barabasi, is a commonly used model for errors in experiments, it is important to remember that there is no experiment for directly measuring the elements of G. Obtaining 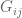
While the exact algorithm (4) for inverting the Barzel-Biham model performs poorly, we found that a widely used shrinkage method based on partial correlation (Schäfer, J. & Strimmer, K. A Shrinkage Approach to Large-Scale Covariance Matrix Estimation and Implications for Functional Genomics, Statistical Applications in Genetics and Molecular Biology 4, (2005)) outperforms the exact algorithm (blue curve in Figure above). This suggests that there is no input for which (4) might be useful. The method is not even ideal for inference from data generated by the model it is based on.
This brings us to the “results” section of the paper. To demonstrate their method called Silencer, Barzel & Barabási ran it on only one of three datasets from the DREAM5 data. They then compared the performance of Silencer to three out of thirty five methods benchmarked in DREAM5. The Barzel-Biham model is for perturbation experiments, but Barzel & Barabási just threw in data from another universe (e.g. mutual information matrices). But lets just go with that for a moment. Their results are shown in the figure below:

Figure 3 from Barzel-Barabási.
The three methods tested potential improvements on are Pearson, Spearman and Mutual Information. Pearson and Spearman rank 16/35 and 18/35 respectively in the DREAM5 benchmarks. There may be some reason why Silencer applied on top of these methods improves performance: in the case where G is a correlation matrix, the path interpretation given by Barzel and Barabási connects the inference procedure to Seawall Wright’s path coefficients (ca. 1920), which in turn suggests an interpretation in terms of partial correlation. However in the case of mutual information, a method that is ranked 19/35 in the DREAM5 benchmarks, there is no statistically significant improvement at all. The improvement is from an AUROC of 0.67 to 0.68. Amazingly, Barzel and Barabási characterize these results by remarking that they “improve upon the top-performing inference methods” (emphasis on top is ours). Considering that the best of these rank 16/35 the use of the word “top” seems, shall we say, unconventional.
We have to ask: how did Barzel & Barabási get to publish a paper in the journal Nature Biotechnology on regulatory network inference without improvement or testing on anything but a handful of mediocre DREAM5 methods from a single dataset? To put the Barzel-Barabási results in context, it is worth considering the standards the Feizi et al. paper were held to. In that paper the authors compared their method to DREAM5 data as well, except they tested on all 3 datasets and 9 methods (and even on a community based method). We think its fair to conclude that significantly more testing would have to be done to argue that Silencer improves on existing methods for biological network inference.
We therefore don’t see any current practical utility for the Barzel-Biham model, except possibly for perturbation experiments in small sub-networks. Even then, we don’t believe it is practical to perform the number of experiments that would be necessary to overcome signal to noise problems.
Unfortunately the problems in Barzel-Barabási spill over into a follow up article published by the duo: Barzel, Baruch, and Albert-László Barabási, “Universality in Network Dynamics.” Nature Physics 9 (2013). In the paper they assume that the local response matrix S has entries that are all positive, i.e. they do not allow for inhibitory interactions. Such a restriction immediately renders the results of the paper, if they are to be believed, moot in terms of biological significance. Moreover, the restrictions on S appear to be imposed in order to provide approximations to G that are unnecessary in light of (5). Given these immediate issues, we suspect that were we to read the Universality paper carefully, it is quite likely this post would have to be lengthened considerably.
These are not the first of Barabási’s papers to package meaningless and incoherent results in Nature/Science publications. In fact, there is a long history of Barabási publishing with fanfare in top journals only to have others respond by publishing technical comments on his papers, in many cases refuting completely the claims he makes. Unfortunately many of the critiques are not well known because they are rejected from the journals where Barabási is successful, and instead find their way to preprint servers or more specialized publications. Here is a partial list of Barabási finest and the response(s):
- Barabasi is famous for the “BA model”, proposed in Barabási and Albert ‘Emergence of Scaling in Random Networks“, Science, Vol. 286 15 October 1999, pp. 509-512. Lada Adamic and Bernardo Huberman immediately refuted the practical applications of the model. Moreover, as pointed out by Willinger, Alderson and Doyle, while it is true that scale-free networks exhibit some interesting mathematical properties (specifically they are resilient to random attack yet vulnerable to worst-case), even the math was not done by Barabási but by the combinatorialists Bollobás and Riordan.
- Barabási has has repeatedly claimed metabolic networks as prime examples of scale-free networks, starting with the paper Jeong et al., “The large-scale organization of metabolic networks“, Nature 407 (2000). This fact has been disputed and refuted in the paper Scale Rich Metabolic Networks by Reiko Tanaka.
- The issue of attack tolerance was the focus of Error and attack tolerance of complex networks by Réka Albert, Hawoong Jeong & Albert-László Barabási in Nature 406 (2000). John Doyle refuted the paper completely in this paper.
- In the paper “The origin of bursts and heavy tails in human dynamics” published in Nature 435 (2005) Barabási pretends to offer insights into the “bursty nature of human behavior” (by analyzing e-mail). In a follow up comment Daniel Stouffer, Dean Malmgren and Luis Amaral demonstrate that the reported power-law distributions are solely an artifact of the analysis of the empirical data and the proposed model is not representative of e-mail communication patterns.
- Venturing into the field of control theory, the paper “Controllability of complex networks” by Liu, Slotine and Barabási, Nature 473 (2011) argues that “sparse inhomogeneous networks, which emerge in many real complex systems, are the most difficult to control, but that dense and homogeneous networks can be controlled using a few driver nodes.” Not so. In a beautiful and strong rebuttal, Carl Bergstrom and colleagues show that a single control input applied to the power dominating set is all that is required for structural controllability of most, if not all networks. I have also blogged about this particular paper previously, explaining the Bergstrom result and why it reveals the Barabási paper to be a control theory embarrassment.
In other words, Barabási’s “work” is a regular feature in the journals Nature and Science despite the fact that many eminent scientists keep demonstrating that the network emperor has no clothes.
Post scriptum. After their paper was published, Barzel and Barabási issued a press release claiming that “their research moves the team a step closer in its quest to understand, predict, and control human disease.” The advertisment seems like an excellent candidate for Michael Eisen’s pressies.

37 comments
Comments feed for this article
February 10, 2014 at 8:50 am
Pedro Mendes (@gepasi)
This work was not even new. In 2002 two very similar methods were published: http://www.ncbi.nlm.nih.gov/pubmed/12142007 & http://www.ncbi.nlm.nih.gov/pubmed/12242336 . Recently they have been applied experimentally: http://www.ncbi.nlm.nih.gov/pubmed/17310240
February 11, 2014 at 5:16 am
pmelsted
The Bollobás, Riordan paper is a beautiful example of coupling graphs. They show that within every preferential attachment graph there is a large sparse random graph and each PA graph has a large part contained inside a sparse random graph. You won’t see any such insight into the underlying random structure in B’s papers.
February 11, 2014 at 2:16 pm
Elchanan Mossel
Critical discussions are good. It’s a shame that “leading” journals do not publish critique, replications (including when things do not replicate) and papers pointing to methodological errors. Given the lack of appropriate forum it is good you publish critique on your blog. Did you ask the authors to take a look and defend themselves?
From a wider perspective, how come everybody knows that the leading science journals are full of bad science and still everybody want to publish there?
February 12, 2014 at 3:22 am
Gareth
“It is not a case of choosing those [faces] that, to the best of one’s judgment, are really the prettiest, nor even those that average opinion genuinely thinks the prettiest. We have reached the third degree where we devote our intelligences to anticipating what average opinion expects the average opinion to be. And there are some, I believe, who practice the fourth, fifth and higher degrees.” (Keynes, General Theory of Employment Interest and Money, 1936). (From Wikipedia.)
As long as we all believe that everybody wants to publish there, we will all want to publish there, right?
February 12, 2014 at 4:19 am
Lior Pachter
Thanks Elchanan.
To answer your question, we submitted a truncated version of this blog post to Nature Biotechnology almost 5 months ago. The two page comment has not yet been decided on by the journal, and my understanding is that a major reason has been the delay of Barzel and Barabási in responding to it. So they did get a chance to respond, but to be honest we just gave up on waiting. Its still possible that Nature Biotechnology will publish our comment and their response. I don’t know, and I don’t particularly care at this point. The authors are of course welcome to post replies in this comments section.
February 18, 2014 at 9:30 pm
Andy Jerkins
Hey Lior,
Can you post this two page comment, like you did for Manolis Kellis’ paper, or is that the same comment?
February 18, 2014 at 11:37 pm
Lior Pachter
We have a separate comment for this paper and it is still under review at Nature Biotechnology. It follows the post closely, and I plan to post it in the near future.
February 13, 2014 at 8:56 am
Guilhem
Thank you for an excellent read. I had myself struggled a lot with the issue of noise when trying their approach in R, and rapidly concluded that the method was simply not usable for any of the biological networks and correlation networks I was interested in. It is astonishing that a method so overly sensitive to noise and multicollinearity is being sold as applicable to biological data.
I think that the following paper by Lima-Mendez and van Helden would be a nice addition to your list of responses to previously published claims:
http://pubs.rsc.org/en/content/articlehtml/2009/mb/b908681a
February 18, 2014 at 12:49 pm
Christos Ouzounis
See also, http://www.ncbi.nlm.nih.gov/pubmed/15496552 our modest contribution to this controversial / trendy subject, years ago.
February 19, 2014 at 4:53 pm
ALB is misleading.
I found the ALB paper very misleading in terms of their claimed contributions. The paper looks pretty but the content is very hollow, not to mention there are technical issues. I showed the paper to one of the students, and he quickly spotted the derivation was nonsense. The journal should clarify why this paper is accepted.
February 26, 2014 at 5:42 am
Yu Xue
Hi, I am a Bioinformatician in China, who is working on computational analysis of post-translational modifications in proteins. My research field is a little bit cross with network analysis. I leave my personal comments below:
1. I really really hate Lior’s blogs, because the three blogs cost me two weeks to understand the major viewpoints. Lior’s great match really scared me. So I have to improve my math for a better understanding, and have to discuss with my friends.
2. Great thanks to write the blogs. My friends and I truly respect you, and what you are doing. Everybody clearly know that you will get no profit from doing so, but you did. You are a great man and a real computational biologists.
3. After two weeks reading and thinking, I wrote three blogs in Chinese, for researchers in China, to introduce Lior’s blogs, as below:
(1) http://blog.sciencenet.cn/home.php?mod=space&uid=404304&do=blog&id=770977
(2) http://blog.sciencenet.cn/home.php?mod=space&uid=404304&do=blog&id=771158
(3) http://blog.sciencenet.cn/home.php?mod=space&uid=404304&do=blog&id=771207
Sorry that I wrote the blogs in Chinese, but I tried my best to keep the major viewpoints of Lior. So Chinese scientists can take a look on it, if you do not want to read English. I will apologize if some misunderstandings were made, because it’s too difficult to me for the math.
It looks like the overhype is quite common in academic society. I am offen stranged that some really poor work can be published in top journals, but noboday dare to stand out. So, the Bioinformatics field should thank for Lior’s criticisms, which is much, much, much helpful for the current and future of the field.
February 27, 2014 at 9:23 am
Junpeng Lao
I am afraid you had made a mistake in your Chinese blog, especially the first one. The scaling parameter is for the eigenvalue but not for the sequence as you explained. You should give a careful read of Lior’s new blog post https://liorpachter.wordpress.com/2014/02/18/number-deconvolution/ for details.
February 28, 2014 at 7:25 am
Jack B.
Lior
You are not the first person to notice that ALB papers were mostly BS.
Same with the Medard and Kellis paper. In fact, the entire literature is full of these kinds of BS. The problem is that people do research to get grants and not the other way around.
But you are the first that had the “balls” to openly announce these findings. I do not have the same amount of courage as you do (and I commend you on that).
March 7, 2014 at 11:15 pm
village-idiot
Yes, there was a short piece in Science not too long ago about this:
http://www.sciencemag.org/content/335/6069/665
But the cottage industry that Barbasi fathered has spun so much out of control that it is unstoppable.
September 27, 2014 at 3:30 pm
Ising Model
This guy should become an NAS and NAE member! He markets his junk quite well.
January 3, 2015 at 3:25 pm
Tom Chou
BTW, comparing oneself to Chandrasekar should be done with care since Edmund Stoner had scooped (and did a more thorough calculation) Chandrasekar but did not get the notoriety for some “reason”. Moreover, Chandrasekar it note cite Stoner, even though the evidence is that he knew about the earlier work.
So, why stop at NAS/NAE….go straight to Stockholm!
January 3, 2015 at 3:50 pm
Tom Chou
Typo…I meant Chandrasekar DID NOT cite Stoner….
January 10, 2015 at 3:44 pm
Nikola
I’m so glad I came across your post today and find out about Barabási’s history of nonsense papers.
He published two closely related papers in Science (http://www.sciencemag.org/content/327/5968/1018) and Nature (http://www.nature.com/nature/journal/v484/n7392/full/nature10856.html#close) on migration and mobility. I am a population geographer and migration researcher and wondered why I could not make sense of a paper published in a prestigious journal. The findings don’t look plausible at all to me, and the model cannot be replicated because the data haven’t been made available…. Anyway, now I know what to think of it.
May 16, 2015 at 10:27 am
virgilio leonardo ruilova castillo
Reblogged this on Virgilio Leonardo Ruilova Castillo.
May 16, 2015 at 10:41 pm
Gatsky
Took the liberty of posting this on hacker news, apologies if this is not appropriate.
The comments are of some interest.
https://news.ycombinator.com/item?id=9555547
May 18, 2015 at 10:03 am
Lior Pachter
Thanks for the post!
May 26, 2015 at 9:46 am
Harry Hab
Delighted to see that I am not the only one who is annoyed by this Barabasi rubbish.
June 15, 2015 at 6:49 am
cbouyio
Reblogged this on CBouyio.
July 3, 2015 at 3:00 am
Takis Konstantopoulos
Lior,
This (and the followup postings) are very much appreciated. I observed the nonsense of Barabasi many times in the past. What puzzles me, however, is that many researchers feel the need to cite his papers. (A quick look at Google shows 22886 citations.) There is a certain “we have to” feeling. People feel they must (for some religious reason?) cite the paper. Recently, I reviewed a paper for a journal with solid mathematical content. They cited the infamous Barabasi-Albert paper stating that they observed a limit result. I told the authors that I took a close look at this paper and found it utterly trivial, with no mathematical content, containing lots of jargon (and, at best, some computer simulations) and, in fact, even the model was not new when the paper was published (1995): the model was known in 1955; see Simon (1955), Biometrica Vol. 42, pp. 425-440.
(The situation reminds me of another in famous paper: On power-law relationships of the Internet topology, by Faloutsos, Faloutsos, and Faloutsos [sic], published in Proc. SIGCOMM ’99, pp. 251-262, and ACM SIGCOMM Computer Communication Review, pp. 251-262. That paper is more modest: 5347 citations (by today). This paper requires a careful dissection too, but it’s already a thing of the past and a waste of our time.)
October 27, 2015 at 9:54 pm
Denise dennis
Ok. almost complete mathematical ignoramus here, but still a teacher, 5th grade. I picked up Linked at B & N. Like science. Opened to pg 98. Bose Einstein condensation. Why here in networks? Read, read, fine, then Pg. 101: !!! Bianconi discovery of incredible similarity of fitness model nodes in networks to energy levels in Bose gas…gosh…suddenly they have done the extraordinary and blended at last the quantum and Newtonian worlds and I’ve heard nothing of it? “…each node in the network corresponds to an energy level in the Bose gas.” All those gas particles at almost 0 Kelvin sink to lowest energy level, new form of matter, and that has what to do with macro world networks and fitness? The rich get richer because the cold all sink down thru energy levels? Help? Keep thinking of similies-apples and toenails or some such. Remembering P. T. Barnum. Elementary school teacher only, me, but afraid I can’t buy this one. Beating Einstein….maybe not this time. Tear me apart, but I really don’t get it. Welcome any explanations-that aren’t too mathematical!
February 10, 2016 at 12:15 pm
3hws
Dear Lior, I find your tone quite rude here. Admittedly, I do not understand your maths discussions; the only thing I can say is that ALB’s network of diseases is highly predictive and inspirational. We are working on an exciting sub-cluster of this, which appears to prove everything true about this new definition of diseases and drug repositioning.
March 8, 2017 at 5:36 am
Daniele
Hi
thanks,
you can add this
https://arxiv.org/abs/1703.00853
March 16, 2017 at 10:11 am
Hedi Hegyi
I agree with Harald regarding the inspirational part. His ideas are quite original and inspirational. I myself cite ALB’s articles in a new paper of mine (coming out soon in Scientific Reports) where I found that schizophrenia genes are actually THE quintessential hub genes interacting with 30 times more genes on average than non-schizophrenia genes. For now m/s is here: http://biorxiv.org/content/early/2016/10/10/080044
May 23, 2017 at 10:08 am
Chao
Dear Professor Pachter,
I am current a Graduate student in New York and I have been following your blog for a long time. In a recent assignment for a course that I am taking, Critical Dissection of Scientific Data, I choose to take up this paper by Barabasi and have read through this blog again. Here is some thoughts I would like to share.
1. As pointed out in http://www.nature.com/nbt/journal/v33/n4/full/nbt.3185.html, the entire mathematical frameworks was developed as Modular Response Analysis (MRA) previously. In fact, their exact solution is exactly your equation (4) here (up to some reformulation) see http://www.pnas.org/content/99/20/12841.abstract, supplemental material. In addition, many following up works has discussed methods of handling noise, see for example, http://essays.biochemistry.org/content/45/161. I am planning to try out their methods on the Erdös-Renyi graph model and see if it works;
2. I attended a talk of one of the developers of MRA, professor Eduardo Sontag (who directed me to your blog). He also pointed out another work of his http://www.sciencedirect.com/science/article/pii/S0006349514004482, which discussed an interesting phenomenon where the assumptions of MRA breaks. I think it is very interesting;
3. With regard to the broader work of Barabasi, professor John Doyle (disclaimer: I am a big fan of him) has claimed in his dropbox video lecture (https://www.dropbox.com/sh/7bgwzqsl7ycxhie/AACL0TVRi5H0k9-r63T1hIyva/VideosSlidesPapers/2.0.OverviewWithNeuroEmphasis?dl=0, ZombiSciIntro) that biological network is in fact, not scale-free, as oppose to what Barabasi has claimed. However it was not clear to me what the explanation is in the video and I can’t find a proper citation yet.
Finally, as long as I am here, I want to thank you for writing these blogs. As a graduate student, I was appalled how Dr. Eric Lander (or someone who claims to be him) reacts to your p-value prize blog and it is now clear to me (after some mental gymnastic) that they were claiming something trivial as significant. So your blog has been a positive impact on my academic life and I believe it is important to read your blog in a sometimes somewhat unfortunate academic environment.
May 23, 2017 at 10:25 am
Lior Pachter
Thanks for your comments and for the links- much appreciated. John Doyle is passionate about the problems of network analysis and has written a lot in the area so I’m not sure which paper is most relevant to the claims in the talk. I know he is still interested in the topic- you should ask him.
May 29, 2017 at 8:03 am
Travis Gibson
Hi Chao, with regard to question 3, I have seen John reference a paper by Tanaka, “Scale-Rich Metabolic Networks” in PRL [http://www.cds.caltech.edu/~doyle/hot/PRL_online.pdf]
Also, the following Nature paper from 2016 may fit into the scope of your class as well https://www.nature.com/nature/journal/v530/n7590/full/nature16948.html [http://barabasi.com/f/687.pdf]. Two preprints pointing out issues with the Nature paper can be found here http://biorxiv.org/content/early/2016/05/31/056218 and here https://arxiv.org/abs/1606.09630
“Here we show the conditions proposed by Gao and collaborators are neither sufficient nor necessary to guarantee that their method works in general, and validity of their results are not independent of the model chosen within the class of dynamics they considered.” – Abstract from the arXiv paper
The underlying issue arises when the original authors assume that the average of the function is the function at the average for explicitly nonlinear maps. This is indeed only true for linear maps.
September 22, 2017 at 8:02 am
Jozsef Haller
The embarrassing fact is that Barabasi got over 100,000 citations to his papers so far. Neither me, nor the authors of this funny contribution equal him in this respect. So, why not throwing mud at him, just for the sake of our ego? The authors’ arguments are partly mathematical in nature. I have nothing to add here. However, I note that there are far more people who refuted Darwin’ evolutionary theory than those who refuted the claims and models of Barabasi. Some maintain that the Earth is flat. Whatever people refute, evolution is the best we can do with the information we have, and the Earth is certainly round. I consider this article a psychological means of addressing threats to the ego. If one cannot grow up to somebody one has to bring him or her down. Nice trial.
(Ps. I am not Barabasi. I am just a biologist newly acquainted with his work, and I was fascinated).
March 2, 2018 at 1:09 am
Nick Halmagyi
Thanks for your interesting and courageous blog! I’m a high energy physicist trying to follow the literature on network theory and stumbled across this page. Regarding point 1. of your summary: I had been pretty stumped on how the BA model from ‘Emergence of Scaling in Random Networks’ could have been quite as innovative as it was promoted to be. I recommend the great review article by Newman https://arxiv.org/abs/cond-mat/0303516v1 specifically section VII where he explains in simple terms how the BA model is a special case of the 1965 citation-network model by Price, which curiously is never cited in the BA paper nor their reviews. I have no horse in this race but the lack of proper accreditation to the Price model by BA made it super confusing for me in trying get a grip on this vast literature. I’m not aware of any other scenario in physics where authors have gotten so much kudos for a “result” which is so clearly a sub-case of a previously published article (Newman points out that both the Price and BA articles were published in Science 🙂 ). As for the complaint that BA were not mathematically precise…I am very supportive of articles being published before being made rigorous, I don’t see any great crime by BA in that regard. The lack of credit given to Price is however really bizarro.
March 9, 2018 at 3:36 pm
rishabh
Hi Lior, your write-up is really interesting. What do you think of multi-task learning for network inference? I found a few examples online:
Click to access 279224.full.pdf
Click to access 279224.full.pdf
February 15, 2019 at 5:34 am
NYT
Albert Lazlo Barabasi, a professor of network science at Northeastern University is in New York Times.
November 13, 2023 at 5:00 pm
SComep
You might be interested in Section 6 of Bollobas’s paper “Mathematical results on scale-free random graphs,” which talks about the famous Barabasi-Albert paper and points out that the model isn’t even cogently formulated on its own terms. https://www.stat.berkeley.edu/~aldous/Networks/boll1.pdf
May 7, 2024 at 10:46 am
Blog do Raphael Winckler
Hi nice reeading your blog