
A few months ago, in July 2019, I wrote a series of five posts about the Melsted, Booeshaghi et al. 2019 preprint on this blog, including a post focusing on a new fast workflow for RNA velocity based on kallisto | bustools. This new workflow replaces the velocyto software published with the RNA velocity paper (La Manno et al. 2018), in the sense that the kallisto | bustools is more than an order of magnitude faster than velocyto (and Cell Ranger which it builds on), while producing near identical results:
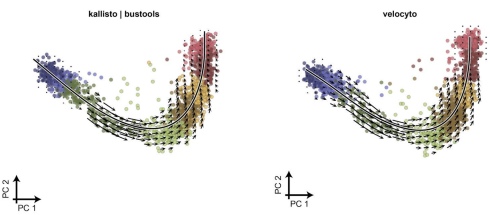
In my blogpost, I showed how we were able to utilize this much faster workflow to easily produce RNA velocity for a large dataset of 113,917 cells from Clark et al. 2019, a dataset that was intractable with Cell Ranger + velocyto.
The kallisto | bustools RNA velocity workflow makes use of two novel developments: a new mode in kallisto called kallisto bus that produces BUS files from single-cell RNA-seq data, and a new collection of new C++ programs forming “bustools” that can be used to process BUS files to produce count matrices. The RNA velocity workflow utilizes the bustools sort, correct, capture and count commands. With these tools an RNA velocity analysis that previously took a day now takes minutes. The workflow, associated tools and validations are described in the Melsted, Booeshaghi et al. 2019 preprint.
Now, in an interesting exercise presented at the Single Cell Genomics 2019 meeting, Sten Linnarsson revealed that he reimplemented the kallisto | bustools workflow in Loompy. Loompy, which previously consisted of some Python tools for reading and writing Loom files, now has a function that runs kallisto bus. It also consists of Python functions that are used to manipulate BUS files; these are Python reimplementations of the bustools functions needed for RNA velocity and produce the same output as kallisto | bustools. It is therefore possible to now answer a question I know has been on many minds… one that has been asked before but not to my knowledge, in the single-cell RNA-seq setting… is Python really faster than C++ ?
To answer this question we (this is an analysis performed with Sina Booeshaghi), performed an apples-to-apples comparison running kallisto | bustools and Loompy on exactly the same data, with the same hardware. We pre-processed both the human forebrain data from La Manno et al. 2018, and data from Oetjen et al. 2018 consisting of 498,303,099 single-cell RNA-seq reads sequenced from a cDNA library of human bone marrow (SRA accession SRR7881412; see also PanglaoDB).
First, we examined the correspondence between Loopy and bustools on the human forebrain data. As expected, given that the Loompy code first runs the same kallisto as in the kallisto | bustools workflow, and then reimplements bustools, the results are the near identical. In the first plot every dot is a cell (as defined by the velocyto output from La Manno et al. 2018) and the number of counts produced by each method is shown. In the second, the correlation between gene counts in each cell are plotted:


The figures above are produced from the “spliced” RNA velocity matrix. We also examined the “unspliced” matrix, with similar results:


In runtime benchmarks on the Oetjen et al. 2018 data we found that kallisto | bustools runs 3.75 faster than Loompy (note that by default Loompy runs kallisto with all available threads, so we modified the Loompy source code to create a fair comparison). Furthermore, kallisto | bustools requires 1.8 times less RAM. In other words, despite rumors to the contrary, Python is indeed slower than C++ !
Of course, sometimes there is utility in reimplementing software in another language, even a slower one. For example, a reimplementation of C++ code could lead to a simpler workflow in a higher level language. That’s not the case here. The memory efficiency of kallisto | bustools makes possible the simplest user interface imaginable: a kallisto | bustools based Google Colab notebook allows for single-cell RNA-seq pre-processing in the cloud on a web browser without a personal computer.
At the recent Single Cell Genomics 2019 meeting, Linnarsson’s noted that Cell Ranger + veloctyto has been replaced by kallisto | bustools:

Indeed, as I wrote on my blog shortly after the publication of Melsted, Booeshaghi et al., 2019, RNA velocity calculations that were previously intractable on large datasets are now straightforward. Linnarsson is right. Users should always adopt best-in-class tools in favor of methods that underperform in accuracy, efficiency, or both. #methodsmatter

4 comments
Comments feed for this article
October 12, 2019 at 6:28 am
Francisco de Abreu e Lima
Thank you Lior, nice discussion!
October 20, 2019 at 10:31 am
YY
Am I the only one confused by the concluding “Linnarsson is right. Users should always adopt best-in-class tools in favor of methods that underperform in accuracy, efficiency, or both”?
Is it mocking or (which seems to me what Lior intended) genuine and something in the text is stilted? Is my ESL failing me?
October 20, 2019 at 8:51 pm
Lior Pachter
Yes, of course it was meant genuinely and sincerely.
November 12, 2019 at 10:26 am
Elizabeth Batory
While this is fascinating, it is also confusing to me – since Python is essentially a shell language for C++ (very little of the computation takes place in pure Python), this is a bit of a silly comparison on one end, and interesting on the other, since I would think that Python would be EXACTLY as fast as C++ for this.