
Continuous-time Markov chain models for DNA mutations on a phylogenetic tree (e.g. the Jukes-Cantor model, the Kimura models, and more generally models of the Felsenstein hierarchy) have the simple and convenient property of multiplicativity. Specifically, if Q is a rate matrix then the associated substitution matrices are multiplicative in the following sense:

This follows directly from the fact that the matrices 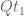
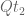

This means that substitutions over a time period 2t are equivalently described as substitutions occurring over a time period t, followed by substitutions occurring afterwards over another time period t.
But what if over the course of time the rate matrix changes? For example, suppose that for a period of time t mutations proceed according to a rate matrix Q, and following that, for another period of time t, mutations proceed according to a rate matrix R? Is it true that the substitutions after time 2t will behave as if mutations occurred for a time 2t according to the (average) rate matrix 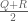
If Q and R commute the answer will be yes, as Qt and Rt will also be commutative and the multiplicativity property will hold. But what if Q and R don’t commute? Is there any relationship at all between 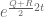

This week I visited Yale University to give a talk in the Center for Biomedical Data Science seminar series. I was invited by Smita Krishnaswamy, who organized a wonderful visit that included many interesting conversations not only in computational biology, but also applied math, computer science and statistics (Yale has strong programs in applied mathematics, statistics and data science, computer science and biostatistics). At dinner I learned from Dan Spielman of the Golden-Thompson inequality which provides a beautiful answer to the question above in the case where Q and R are symmetric. The theorem is a trace inequality for Hermitian matrices A and B:

This inequality is well known in statistical mechanics and random matrix theory but I don’t believe it is known in the phylogenetics community, hence this post. The phylogenetic interpretation of the pieces of the Golden-Thompson inequality (replacing A with Qt and B with Rt) is straightforward:
- The matrices
- The product
is the substitution matrix corresponding to mutations occurring with rate matrix Q for time t followed by rate matrix R for time t.
- The matrix
is the substitution matrix for mutations occurring with rate
- Since the trace of a substitution matrix is the probability that there is no transition, or equivalently the probability that a change in nucleotide does not occur, the Golden-Thompson inequality states that for two symmetric rate matrices Q and R, the probability of a substitution after time 2t is higher when mutations occur first at rate Q for time t and then at rate R for time t, than if they occur at rate
In other words, rate changes decrease the expected number of substitutions in comparison to what one would see if rates are constant.
The Golden-Thompson inequality was discovered independently by Sidney Golden and Colin Thompson in 1965. A proof is explained in an expository blog post by Terence Tao who heard of the Golden-Thompson inequality only eight years ago, which makes me feel a little bit better about not having heard of it until this week! It would be nice if there was a really simple proof but that appears not to be the case (there is a purported one page proof in a paper titled Golden-Thompson from Davis, however what is proved there is the different inequality 
There is considerable interest in evolutionary biology in models that allow for time-varying rates of mutation, as there is substantial evidence of such variation. The Golden-Thompson inequality provides an additional insight for how mutation rate changes over time can affect naïve estimates based on homogeneity assumptions.

The Felsenstein hierarchy (from Algebraic Statistics for Computational Biology).

3 comments
Comments feed for this article
October 9, 2018 at 10:12 am
Daniel Weissman
Interesting post! I didn’t understand all of it. Three things in particular I’m wondering about:
1. (Kind of silly.) Is it obvious that the increase in the expected number of sites without a substitution implies that the expected number of substitutions decreases? If we’re talking codons so that multiple substitutions could be observed at a single site, is it possible for the former to be true without the latter?
2. Should the bolded sentence be weakened, since it only applies to symmetric rate matrices? Even if we ignore selection, AT-GC mutation bias would create asymmetries. I think it’s important to allow these asymmetries to account for observed changes in base composition and codon bias.
3. I think I’m missing something, because it’s not obvious to me that the trace is the probability that no substitution occurs. There’s a trivial terminological quibble about reversions, but ignoring that, it seems like it should only be true when the initial distribution of states is uniform.
October 20, 2018 at 12:36 am
Saket Choudhary
Hi Lior,
Thanks for writing this. We happened to cover this in EE-546 (https://web-app.usc.edu/soc/syllabus/20183/30401.pdf) but never covered the proof. I happened to dig in a bit deeper and came across a more intuitive proof from 2014 by David Sutter, Mario Berta and Marco Tomamichel: https://arxiv.org/pdf/1604.03023.pdf
A slightly elaborated version (I had difficulty figuring out a few of the lemmata and tried to detail it out):
https://drive.google.com/file/d/1vUAv0TMiAN-wnwN6jeWwQSqJS0ucxnWv/view?usp=sharing
January 19, 2020 at 12:06 am
Lior Pachter
Thanks. For interested readers I’m linking to your arXiv preprint that followed the comment above:
https://arxiv.org/abs/1811.00544