
On the 27th March, 2012 the Foreign Currency Department of the Central Bank of Iceland raided the offices of Samherji hf., a fishing company in Iceland. The Office of the Special Prosecutor was concerned that Sammherji hf. might be shifting profits in Iceland to its overseas subsidiaries by underselling its fish. In doing so it would be recording losses in Iceland while sheltering profits abroad thereby violating currency exchange (and possibly other) laws. The raid was immediately addressed by the company in a press release; for a complementary perspective see the article by journalist Ingi Freyr Vilhjálmsson in Icelandic/Google English translation.
Two months later, in a sharply worded rebuttal, the company argued that the reported discrepancies in prices between Samherji hf. and the market were flawed. Specifically, they argued, the Central Bank had incorrectly averaged numbers (Google English translation) resulting in a Simpson’s paradox! Was this a valid argument? Truly an example of Simpson’s paradox? Or a sleight of hand?
Simpson’s paradox (better referred to as Simpson’s reversal) is a mathematical inequality that can arise in the comparison of averages with raw data. It is known as a “paradox” because the inequality can appear to be unintuitive (to those who have poor statistical intuition, i.e. most of us). What makes it particularly perilous is that it can lurk disguised in the most unexpected places. Not only (allegedly) in the Central Bank of Iceland, but also, it turns out, in many comparative genome analyses…
Original sin
The mouse genome was published in December 2002, and in the publication was the following figure (Figure 25 from the mouse genome paper):

In the review Sixty years of genome biology by Doolittle et al. (2013) highlighting key advances in genome biology since the publication of the structure of the double helix in 1953, Chris Ponting singles out panel (a) as being of historical significance describing it as “a wonderful visual guide to the most important features of mammalian genes” and explaining that “collapsing levels of sequence conservation between thousands of mouse and human orthologs into one metagene, .. showed how, from a common sequence over 90 million years ago, mutation has etched away intronic sequence whilst selection has greatly preserved the exons, particularly toward their boundaries”. He is right, of course, in that the figure demonstrated, for the first time, the power of “genome-wide” comparative molecular biology and led to concerted efforts to characterize functional regions in the genome by aggregation of genome-wide data. Figures analogous to the mouse genome paper 25a are now a fixture in many genomics papers, with % sequence identity being replaced by data such as % methylated, % accessible, etc. etc.
In the recent paper
M. Singer and L. Pachter, Controlling for conservation in genome-wide DNA methylation studies, BMC Genomics, 16:240
my former Ph.D. student Meromit Singer (now a postdoc in the Regev lab at the Broad Institute) and I explain why in some cases the type of “meta-analysis” underlying Figure 25a, now a fixture in many genomics papers, can be misleading. We focused on DNA methylation studies, which I will explain, but first some elementary algebra…
Simpson’s paradox
According to the English Oxford Dictionary, one of the definitions for the word paradox is “A statement or proposition which on the face of it seems self-contradictory, absurd, or at variance with common sense, though, on investigation or when explained, it may prove to be well-founded”, and it is this sense of the word that is relevant in what has become known as “Simpson’s paradox”. The “paradox”, is just the following fact:
Given numbers 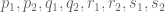



The geometry is that in the figure below (from wikipedia), even though each of the red vectors has a slope greater than its corresponding blue vector, the sum of the red vectors has a slope less than the sum of the blue vectors:

This reversal, nothing more than a simple algebraic curiosity, can sometimes underlie qualitative changes in the results of data analysis after averaging. This is because ratios or rates can be recast as slopes of vectors while averages correspond to sums of vectors. A famous example is described in the classic paper on sex bias in graduate admission at UC Berkeley, Bickel et al. discuss the issue of pooling data, and how it can affect conclusions about bias. The main point of the paper, namely that Simpson’s paradox can emerge in analyzing admissions data, is illustrated in the following toy example (Figure S1b) from our paper, which was constructed to facilitate understanding the effect in the context of genomics:

The figure shows (hypothetical) applications and admissions results for eight males and eight females at twelve departments in a university. A “0” indicates an applicant applied and was rejected, a “1” that the applicant applied and was accepted, and a grey box indicates that the applicant did not apply. In all departments the admissions rates are the same for males and females (0% for both males and females in departments 1–4 and 100% for both males and females in departments 5–12, best understood as the ratio, in each row, of the sum of the numbers divided by the number of non-grey squares). Yet the acceptance rate for all males is 66.7% vs. 33.3% for females (for each column, sum of the numbers divided by the number of non-grey squares as displayed in the plot underneath the table). This is a strange effect at first glance… somehow grouping males and females together leads to an appearance of bias, whereas each department is admitting students without regard to gender (the paper by Bickel et al. discusses real data from UC Berkeley admissions in 1973 where this effect resulted in statistically unjustified claims of bias).
Looking at the table it is clear that the reason for the discrepancy between departmental admission rates and overall admissions rates is due to the extra grey boxes in the female table. In this example (and in the 1973 UC Berkeley admissions) females applied to departments that are harder to get in to. In the example above the male and female departmental admissions rates were equal, but it is not difficult to construct examples (again, they occurred in the 1973 UC Berkeley data) were a reversal happens, i.e. admissions rates at the departmental level are opposite to those when aggregated. This is what is known as “Simpson’s paradox”.
That the issue is relevant in the genomics meta-analysis context can be seen by replacing “males” and “females” with by genomic features, e.g. “introns” and “exons”, and “departments” with different genomic instances of the features. For example, in the case of Figure 25a from the mouse genome paper, “departments” correspond to different instances of introns/exons in the genome, and “0”s to “not conserved” and “1”s to “conserved”. Interestingly, in the genomics meta-analysis context it is the plot (column averages in the table above) that is always the displayed “result”, and this makes the “Simpson’s effect” more subtle to detect or to identify than in standard settings where the table itself is revealed.
What is the equivalent of “females apply to departments that are harder to get into”? Consider the human-mouse plot in Figure 25a of the mouse genome paper. In some genes some bases are not conserved between human and mouse, and this is far more likely to happen in introns than exons. In fact, this is shown in Figure 25a (light blue curve labeled “aligning”). Hence the grey boxes of missing data, reflecting the fact that introns are less conserved than exons, and therefore in alignments they have not only more mismatches, but also indels.
In our paper, we considered the case of DNA methylation measurements, where “0” referred to unmethylated and “1” to methylated. Since DNA methylation can only be measured at CpGs, there is again, the issue of missing data. And as it turns out, the missing data is associated with feature type, so that genome-wide averaging can be a huge problem.
DNA Methylation
In the course of working on her Ph.D. on Statistical algorithms in the study of DNA methylation, Meromit worked on identifying and understanding DNA methylation inside genes, and in that context was interested in Figure 4 from the paper Dynamic changes in the human methylome during differentiation by L. Laurent et al., Genome Research, 20 (2010), p 320–331. The figure describes “average distribution of DNA methylation mapped onto a gene model”, and is reproduced below:

Figure 4 from L. Laurent et al., Genome Research 2010.
Figure 4c plays a major role in the paper, supporting the following sentence from the abstract: “Exons were more highly methylated than introns, and sharp transitions of methylation occurred at exon–intron boundaries, suggesting a role for differential methylation in transcript splicing.” Throughout the paper, there is extensive discussion of “sharp steps”, not only at exon-intron boundaries but also transcription start sites (TSS) and transcription termination sites (TTS), with speculation about the regulation such methylation differences might reflect and contribute to. Of particular note is the following paragraph about Figure 4c:
“A downward gradient was seen going across exons from 5′ to 3′, while an upward gradient of DNA methylation was seen traveling from 5′ to 3′ across introns (Fig. 4C). While this is the first report on the splice junction methylation spikes, recent reports show that the intron–exon boundaries also appear to be marked by gradients in chromatin features, including nucleosomes (Schwartz et al. 2009) and the H3K36me3 histone mark (Kolasinska-Zwierz et al. 2009). Taken together, our data suggest that coupling of transcription and splicing may be regulated by DNA methylation as well as by other epigenetic marks.”
Meromit focused on the italicized sentence (emphasis mine). Looking at the figure, she noticed that the 5′ ends of exons seemed to be much more highly methylated than the 3′ ends. This, and the discussion in the paper, led her to think that indeed DNA methylation may be functional within transcripts. But given the sparsity of CpGs (for one thing, it turned out that the spikes at the intron-exon boundaries were due to an almost complete lack of CpGs there), and noting that CpGs must be present for DNA methylation to be measured, she decided to examine whether there was in fact stronger conservation of CpGs at the 5′ end of exons. However, when reanalyzing the data, she discovered that the reason for the reported difference in methylation was that because intragenic junctions were examined, the 3′ ends of exons were on average closer to the promoter than the 5′ ends, and therefore the difference in methylation was simply reflecting the fact that many first exons are unmethylated due to “leakage” from the promoter. This, in turn, got her thinking about the interplay between DNA methylation, conservation of sequence, and bias that may occur during the averaging of data.
One of my favorite figures from our paper is our Figure 3, which illustrates what can and does go wrong in naïve genome-wide averaging of methylation data at functional boundaries:

The leftmost column shows the result of naïve plotting of column averages for %methylated CpGs in simulations where the methylation states from real data was shuffled randomly across features (while maintaining the locations of missing data due to absent CpGs). The “sharp steps” reported in Laurent et al. are evident despite the fact that there is no actual difference in methylation. The next column (second from left) is the naïve plot generated from real data (4852 5′ UTR-coding junctions, 20,784 mid-gene intron-exon junctions and 21,408 CpG island junctions from genome-wide bisulfite data generated by Lister et al., 2008). Evidently the “sharp steps” look very similar to the simulated scenario where there is no actual methylation difference across the boundaries. The “Corrected via COMPARE” column shows what happens when the bias is removed via a correction procedure that I describe next. In the case of UTR-coding junctions, the “sharp step” disappears completely (this is confirmed by the analysis shown in the final column, which consists of simply throwing away rows in the data table where there is missing data, thereby removing the possible Simpson’s effect but at the cost of discarding data). Our analysis also shows that although there is some differential methylation (on average) at intron-exon boundaries, the effect is smaller than appears at first glance.
Without going into too much detail, it is worth noting that the underlying problem in the methylation case is that deamination leads to methylated CpGs converting to TpGs, so that this is a correlation between methylation and missing data. This is analogous to the problem in conservation analysis, where missing data (due to indels) is correlated with less conservation (i.e. more mismatches). Our paper delves into the details.
Although the analysis in Laurent et al. is problematic, the intention of this post is not to single out the paper; it just happens to be the publication that led us to the discovery of Simpson’s effect in the context of genome-wide averaging. In fact, in the preparation of our paper, we identified 40 published articles where naïve averaging of genome-wide methylation data was displayed in figures (the same issue undoubtedly affects other types of genome analysis as well, e.g. conservation analysis as in Figure 25a from the mouse paper, and we make this point in our paper but do not explore it in detail). In some cases the interpretation of the figures is central to the claims of the paper, whereas in other cases the figures are secondary to the main results. Also sometimes correction of Simpson’s effect may lead to a (small) quantitative rather than a qualitative change in interpretation; we have not examined each of the 40 cases in our table in detail (although are happy to send the table upon request). What we recommend is that future studies implement a correction when averaging genome-wide data across features. To assist in this regard, Meromit developed software called COMPARE:
Imputation of missing data
There are basically three ways to get around the problem discussed above:
- Don’t average the data in your tables.
- Discard rows with missing entries.
- Impute the missing values.
The problem with #1 is that summaries of data in the form of plots like Figure 25a from the mouse genome paper are a lot more fun to look at than tables with thousands of rows. The workflows for #2 and #3 above are shown in our Figure 4:

However the problem with #2 is that at least in the case of DNA methylation, a lot of data must be discarded (this is evident in the large boxes in the final column of our Figure 3; see above). Imputation is interesting to consider. It is essentially the statistical problem of guessing what the methylation state in a specific position of a DNA sequence would be if there was a CpG at the site. The idea is to perform matrix completion (in a way analogous to the methods used for the Netflix prize); details are in our paper and implemented in software called COMPARE. It turns out that imputation of epigenetic state is possible (and can be surprisingly accurate):

The plots show analysis with COMPARE, specifically ten-fold cross-validations for 5’UTR, coding, intronic and exonic regions, at the corresponding junctions analyzed in Figure 3. Smoothing was used to display the large number of data points. In each plot n is the number of data points in the matrix and the regression line is shown in dark blue where s is its slope, and v is the additional amount of variance in the data explained by the regression line relative to a random line. Our results are consistent with those recently published in Ernst and Kellis, 2015, where the tractability and application of imputation of epigenetic states is discussed.
With COMPARE, it is possible to generate corrected plots (see column 3 in our Figure 3 above) that provide a clearer quantitative picture of the data. A reanalysis of intron-exon junctions with COMPARE is interesting. As shown in Figure 3 there is a substantially reduced effect in human (approximately a factor of two) in comparison to naïve averaging. What is interesting is that in Arabidopsis correction with COMPARE hardly changes the result at all (Figure S6):

The upshot is that after correction in both human and Arabidopsis, a correction that affects human but not Arabidopsis, the differences in DNA methylation at intron-exon boundaries are revealed to be the same.
Despite the obvious importance of imputation in this context, we discovered in the course of trying to publish our paper that some biologists are apparently extremely uncomfortable with the idea (even though SNP imputation has become standard for GWAS). Moreover, it seemed that a number of editors and reviewers felt that the reporting and characterization of abundant bias affecting a large number of publications was simply not of much interest. The publication process of our paper was one of the hardest of my career; we went through several journals and multiple review stages, including getting rejaccted (not a typo!). Rejacction is the simultaneous rejection and acceptance of a paper. To wit, an editor wrote to say that “unfortunately, we feel that as it stands the manuscript…cannot [be] consider[ed].. in its current form [but] we have taken the liberty of consulting the editors of [another journal], and they have informed us that if you decided to submit your manuscript to [them], they would only ask for a quick overview.” In other words, rejected, but accepted at our sister journal if you like. We were rejaccted twice (!) bringing to mind a (paraphrasing) of Oscar Wilde: to be rejected once may be regarded as misfortune; twice looks like carelessness. At the end, we were delighted to publish our paper in the open access journal BMC Genomics, and hope that people will find it interesting and useful.
On fishing
Returning to the claims of Samherji hf. about the raid, there is a point to discussing Simpson’s effect in the context of comparing fish prices. The argument being made, is that the Central Bank of Iceland averaged across different weight classes of fish in computing the ratios “price per kilogram” (true) and that it would be better to instead compute weighted averages according to the weight of the fish sold (unclear). Specifically, while Simpson’s reversal can occur in the setting Samherji hf. is referring to, it is not at all clear from the data presented that this is what happened (I would check, but it would require knowledge of prices both of Samherji hf. and of competitors, and also tables that are not in pdf as in the attachments provided). Instead, all that Samherji hf. is saying is that they prefer a weighted averaging in computing price per kilogram, a procedure that, a priori, is by no means clearly “better” than naïve averaging for issues the Central Bank was investigating. After all, just because small fish might sell for different prices than large fish, it is not clear that such fish should be discounted in computing average prices per kilogram. Moreover, it is not at all apparent why Simpson’s paradox is relevant if the matter under consideration is whether or not to weight results when averaging. This is all to say that Simpson’s paradox can not only be subtle to detect, but also subtle to interpret if and when it does occur. One of the notorious abuses of Simpson’s paradox is its use by Republicans in claiming that more of them voted for the 1964 civil rights act than Democrats. It turns out that while this is true, the opposite is true as well.
And so it can be with genomics data. Be careful when you fish.
Disclosures: I was an author on the mouse genome paper. I am employed by UC Berkeley. I am the brother-in-law of the journalist mentioned in the opening paragraph. I eat fish.

9 comments
Comments feed for this article
July 13, 2015 at 8:24 am
Anon
Very interesting. I wonder if there’s an extension of this problem to Chip-Seq data. Many, many papers tend to show aggregated ChIP signal around a feature of interests. I wonder if issues of mappability, library depth, etc. may have a similar effect to missing data in methylation data, when one seeks to aggregate. Do you know of any works which have explored this issue (if there is one)?
July 13, 2015 at 10:39 am
Anshul Kundaje
Nice post. We discussed related issues with global averaging of ChIP-seq and MNase-seq data around landmark sites like TSSs and TFBSs in this paper. http://m.genome.cshlp.org/content/22/9/1735.full
July 13, 2015 at 10:42 pm
Ming Tang (@tangming2005)
for ChIP-seq, in addition to the average plot, a heatmap that with each region in each row should make it more clear to compare (although not quantitatively). a box-plot (or a histogram) is better in this case . I am really uncomfortable averaging the signal, as a single value (mean) is not a good description of the distribution.
July 14, 2015 at 11:04 am
Meromit Singer
With respect to ChIP-Seq data and the like, it would be important to consider whether there are differences between the probability of false-negatives (or false-positives) and some feature like the underlying sequence. For example, if conserved DNA sequences are less probable to be bound by protein X but are also more probable to be sequenced for some reason, that could be a problem.
In contrast to bisulfite data profiling DNA methylation, in ChIP-Seq the generation of data itself from a location is indicative of binding, which makes decoupling of “missing data” and “negative data” more involved. In the DNA methylation setting, “missing data” (no CpG at a genomic site so no data of the potential methylation state had it been a CpG) is different from “negative data” (data collected at a CpG site but no DNA methylation detected). This decoupled setting enabled us to isolate the Simpson’s effect for characterization and correction, and I think it will be interesting to see how the Simpson’s effect can be detected and treated in cases in which “no data” at a location is an indication of either: “missing data” (e.g. protein X bound but region not sequenced) or “negative data” (protein X not bound at location).
@Anshul – thanks for the paper ref! Indeed, an additional important issue with averaging is that one could be looking at the aggregation of several (possibly very distinct) clusters. Another thing we should all keep in mind if we choose to make such plots..
July 13, 2015 at 12:23 pm
GenomeHunter
Interesting point. As you mentioned, Ersnt and Kellis also try to impute the missing epigenomic marks using the available intra- and inter-cell type information. It would be interesting to quantitatively compare COMPARE with their proposed method.
What I don’t like about Ersnt and Kellis’ work is the complete ignorance of the sequence context. I don’t think this problem is same as Netflix’s matrix completion problem, as ordering of the movies/raters does not matter. While in any sort of genomic data there are (strong) proximal and distal spatial correlations between genomic features. Moreover, it is very likely that these correlations are sequence (context or even motif) dependent.
GH
July 13, 2015 at 5:41 pm
LLL
Just out of curiosity, imputation is one way to fill in the missing data. Do you think another approach inverse probability weighted estimators will also applicable here? My understanding is that IPW estimators are in general easier to compute, the extension of which enjoy the doubly robustness property.
July 14, 2015 at 9:08 pm
sparc
As a non-biostatistician who worked with PBMNCs I wonder why nobody complained about the following bit in the paper’s abstract:
because PBMNCs are just not only monocytes.
May 22, 2016 at 8:43 pm
Andy Jerkins
Hey All,
Out of curiosity, does the result presented in the first figure (i.e., conservation across the gene body) suffer from the issue you described here? Does the magnitude of effect change?
May 23, 2016 at 6:39 am
Lior Pachter
Yes. For starters we looked at the relationship between the number of alignments and conservation scores, and as you might expect there is a strong correlation (regions with strong conservation scores have many alignments). This is analogous to the absence of CpGs in regions where CpGs are highly methylated. We felt that a complete and careful analysis of the magnitude of the resulting effect was beyond the scope of our paper, but it certainly does matter.
By the way, I recently read Jordan Ellenberg’s book “How not to be wrong” in which he tells the story of Abraham Wald and the “missing bullet holes”. See, e.g. http://thepenguinpress.com/2014/05/new-nonfiction-how-not-to-be-wrong-the-power-of-mathematical-thinking-by-jordan-ellenberg/
This is exactly the same thing that is going on in conservation analysis (or methylation studies).