
This is the second in a series of three posts (part1, part3) on two back-to-back papers published in Nature Biotechnology in August 2013:
- Baruch Barzel & Albert-László Barabási, Network link prediction by global silencing of indirect correlations, Nature Biotechnology 31(8), 2013, p 720–725. doi:10.1038/nbt.2601
- Soheil Feizi, Daniel Marbach, Muriel Médard & Manolis Kellis, Network deconvolution as a general method to distinguish direct dependencies in networks, Nature Biotechnology 31(8), 2013, p 726–733. doi:10.1038/nbt.2635
The Barzel & Barabási paper we discussed in yesterday’s blog post is embarrassing for its math, shoddy “validations” and lack of results. However of the two papers above it is arguably the “better” one. That is because the paper by Soheil Feizi, Daniel Marbach, Muriel Médard & Manolis Kellis, in addition to having similar types of problems to the Barzel & Barabási paper, is also dishonest and fraudulent. For reasons that I will explain in the third and final post in this series, we (Nicolas Bray and I) started looking at the Feizi et al. paper shortly after it was published early in August 2013. This is the story:
Feizi et al.‘s paper describes a method called network deconvolution that in their own words provides “…a systematic method for inferring the direct dependencies in a network, corresponding to true interactions, and removing the effects of transitive relationships that result from indirect effects.” They claim that the method is a “foundational graph theoretic tool” and that it “is widely applicable for computing dependencies in network science across diverse disciplines.” This high brow language makes network deconvolution sounds very impressive, but what exactly is the method?
Feizi et al. would like you to believe that the method is what is described in their Figure 1, part of which is shown below:

This is a model for networks represented as matrices. In this model an unknown matrix 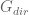
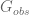
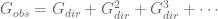
The matrix

Unfortunately, many matrices cannot be decomposed as an infinite sum of powers of some matrix as in equation (1), so equation (2) cannot be applied directly to arbitrary data matrices. The only mention of this issue in the main text of the paper is the statement that “This [eigenvalue] assumption can be achieved for any matrix by scaling the observed input network by a function of the magnitude of its eigenvalues.” This is true but incoherent. There are an infinite number of scalings that will satisfy the assumption, and while the authors claim in their FAQ that “the effect of linear scaling on the input matrix is that … it does not have an effect” this is obviously false (also if it has no effect why do they do it?). For example, as the scaling goes to zero,
What the authors have actually done with their seemingly innocuous aside is fundamentally change their model: now instead of (1),
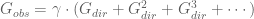
The problem with this model is that given 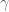
In the video below, recorded at the Banff workshop on Statistical Data Integration Challenges in Computational Biology: Regulatory Networks and Personalized Medicine (August 11–16, 2013), you can see an explanation of network deconvolution by the corresponding author Kellis himself. The first part of the clip is worth watching just to see Kellis describe inverting a matrix as a challenge and then explain the wrong way to do it. But the main point is at the end (best viewed full screen with HD):
Manolis Kellis received his B.S., M.S. and Ph.D degrees in computer science and electrical engineering from MIT, so it is hard to believe that he really thinks that solving (2), which requires nothing more than a matrix inversion, must be accomplished via eigendecomposition. In fact, inverting a 2000 x 2000 matrix in R is 50% slower using that approach. What is going on is that Kellis is hiding the fact that computation of the eigenvalues is used in Feizi et al. in order to set the scaling parameter, i.e. that he is actually solving (3) and not (2). Indeed, there is no mention of scaling in the video except for the mysteriously appearing footnote in the lower left-hand corner of the slide starting at 0:36 that is flashed for 2 seconds. Did you have time to think through all the implications of the footnote in 2 seconds, or were you fooled?
While it does not appear in the main text, the key scaling parameter in network deconvolution is acknowledged in the supplementary material of the paper (published with the main paper online July 14, 2013). In supplementary Figure 4, Feizi et al. show the performance of network deconvolution with different scaling parameters (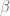
 Figure S4, July 14, 2013.
Figure S4, July 14, 2013.
In the words of the authors, the point of this figure is that “…choosing
We inquired with the authors and were surprised to discover that while 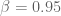
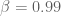

Although it may be hard to believe, this is where the story gets even murkier.
Perhaps as a result of our queries, the scaling parameters were publicly disclosed by Feizi et al. in a correction to the original supplement posted on the Nature Biotechnology website on August 26, 2013. The correction states “Clarification has been made to Supplementary Notes 1.1, 1.3, 1.6 and Supplementary Figure 4 about the practical implementation of network deconvolution and parameter selection for application to the examples used in the paper. ” But Supplementary Figure 4 was not clarified, it was changed, a change not even acknowledged by the authors, let alone explained. Below is Figure S4 from the updated supplement for comparison with the one from the original supplement shown above.
 Figure S4, August 26, 2013.
Figure S4, August 26, 2013.
The revised figure is qualitatively and quantitatively different from its predecessor. A key point of the paper was changed- the scaling parameter
1. If the authors believed their original figure and their original statement that
2. When and how was the July 16th Figure S4 made?
Manolis Kellis declined to answer question 1 and by not acknowledging the figure change in the correction did not permit the readers of Nature Biotechnology to ask question 2.
Clearly we have fallen a long way from the clean and canonical-seeming Figure 1. We wish we could say we have reached the bottom, but there is still a ways to go. It turns out that “network deconvolution” is actually neither of the two models (2) and (3) above, a fact revealed only upon reading the code distributed by the authors. Here is what is in the code:
- affinely map the entries of the matrix
- set the diagonal of the matrix to 0,
- threshold the matrix, keeping only the largest
fraction of entries,
- symmetrize the matrix,
- scale the matrix so that
- apply formula (2),
- affinely map between 0 and 1 again.
The parameter 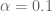
It is hard to say what steps 1–7 actually do. While it is impossible to give a simple analytic formula for this procedure as a whole, using the Sherman-Morrison formula we found that when applied to a correlation matrix C, steps 1, 2, 4, 6 and 7 produce a matrix whose ijth entry is (up to an affine mapping) 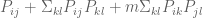

When Feizi et al. was published in August 2013, they set up a companion website at http://compbio.mit.edu/nd/ where the code and data was made available (the data is no longer available at the site). On December 4, 2013, the authors updated the website. While there used to be one version of the program, there are now two different versions, one specifically for use on regulatory networks where the default values for the parameters are 

To summarize, “network deconvolution”, as best as we can currently tell, consists of steps 1–7 above with the parameters:
- DREAM5 challenge:
and
- Protein network:
.
- Co-authorship:
and
The description of the method in the main text of the paper is different from what is in the online methods, which in turn is different from what is in the supplement, which in turn is different from what is actually in each of the (two) released programs, namely the ad-hoc heuristic steps we have described above.
Having said all of that, one might be curious whether the “method” actually works in practice. Sometimes heuristics work well for reasons that are not well understood (although they are usually not published in journals such as Nature Biotechnology). Unfortunately, we have to disclose that we don’t know how the method performs on the datasets in the paper. We tried really hard to replicate the results of the paper, running the software on the provided input matrices and comparing them to the distributed output matrices (available on the NBT website). Despite our best attempts, and occasionally getting very close results, we have been unable to replicate the results of the paper. This bothers us to such an extent that I, Lior Pachter, am hereby offering $100 to anyone (including Feizi et al.) who can provide me with the code and input to run network deconvolution and produce exactly the figures in the paper (including both versions of Figure S4) and the output matrices in the supplementary data.
Because we couldn’t replicate the results of the paper, we decided to examine how FK performs in one case for which Feizi et al. originally claimed (Supplement version July 14 2013) that their method is optimal. They stated “if the observed network is a covariance matrix of jointly Gaussian variables, ND infers direct interactions using global partial correlations”. This claim was deleted without mention in the updated supplement, however even if network deconvolution is not optimal for inferring a network from a correlation matrix (presumably what they meant in the original statement), the claimed universality suggested in the paper implies that it should be very good on any input.

The figure above shows a comparison of the Feizi et al. method (FK) with the various beta parameters used in their paper to regularized partial correlation, the “geometric root” (equation (2)) and the naïve method of simply selecting the top entries of the correlation matrix. In order to test the method we performed 40 simulations of sampling 500 observations from a random Gaussian graphical model with 1000 variables and an edge density of 5% to ensure the graph was connected yet sparse. Performance was assessed by comparing the ranking of the edges to the true edges present in the model and computing the area under the corresponding ROC curve. As a benchmark we applied partial correlation structural inference based on a James-Stein shrinkage estimation of the covariance matrix (a standard approach to gene regulatory network inference, described in Schäfer, J. & Strimmer, K. A Shrinkage Approach to Large-Scale Covariance Matrix Estimation and Implications for Functional Genomics. Statistical Applications in Genetics and Molecular Biology 4, (2005)).
That FK does better than the most naïve method and yet worse than a method actually designed for this task is perhaps to be expected for a heuristic method that is based on a metaphor (one that dates back to Seawall Wright’s work on path coefficients in the 1920s- although Wright used (1) in the restricted setting of directed acyclic graphs where it makes sense). It is possible that in some contexts, FK will make things somewhat better. However what such contexts might be is unclear. The only guidance that Feizi et al. provide on the assumptions needed to run their method is that they state in the supplement that the model assumes that networks are “linear time-invariant flow-preserving operators”. While it is true that individual parts of that phrase mean something in certain contexts, the complete phrase is word salad. We wonder: is co-authorship flow preserving?
To conclude: Feizi et al. have written a paper that appears to be about inference of edges in networks based on a theoretically justifiable model but
- the method used to obtain the results in the paper is completely different than the idealized version sold in the main text of the paper and
- the method actually used has parameters that need to be set, yet no approach to setting them is provided. Even worse,
- the authors appear to have deliberately tried to hide the existence of the parameters. It looks like
- the reason for covering up the existence of parameters is that the parameters were tuned to obtain the results. Moreover,
- the results are not reproducible. The provided data and software is not enough to replicate even a single figure in the paper. This is disturbing because
- the performance of the method on the simplest of all examples, a correlation matrix arising from a Gaussian graphical model, is poor.
In academia the word fraudulent is usually reserved for outright forgery. However given what appears to be deliberate hiding, twisting and torturing of the facts by Feizi et al., we think that fraud (“deception deliberately practiced in order to secure unfair or unlawful gain”) is a reasonable characterization. If the paper had been presented in a straightforward manner, would it have been accepted by Nature Biotechnology?
Post scriptum. After their paper was published, Feizi et al. issued some press releases. They explain how original and amazing their work is. Kellis compliments himself by explaining that “Introducing such a foundational operation on networks seems surprising in this day and age” and Médard adds that “the tool can be applied to networks of arbitrary dimension.” They describe the method itself as a way to have “…expressed the matrix representing all element correlations as a function of its principal components, and corresponding weights for each component” even though principal components has nothing to do with anything in their paper. As with Barzel-Barabási, this seems worthy of nomination for the Pressies.
Post post scriptum: In September 2013, we submitted a short commentary to Nature Biotechnology with many of the points made above. While it was rejected four months later, we really really wish we could share the reviews here. Unfortunately they were sent to us with a confidentiality restriction, though having said that, any of the reviewers are hereby invited to guest post on network science on this blog. We were disappointed that Nature Biotechnology would not publish the commentary, but not surprised. Frankly, it would have been too much to expect them and their reviewers to unravel the layers of deception in Feizi et al. in the initial review process; it took us an extraordinary amount of time and effort. Having published the paper, and without a clear path to unpublication (retraction is usually reserved for faking of experimental data), there were presumably not many options. The question is whether anything will change. Feizi et al. apparently have more Nature Biotechnology papers on the way as evidenced by an extraordinary declaration on Soheil Feizi’s publication page (this is a pdf of the version when this blog was posted; Feizi changed his site an hour later) where the publication venue of their next paper appears to have been preordained:
S. Feizi, G. Quon , M. Mendoza, M. Medard, M. Kellis
Comparative Analysis of Modular Integrative Regulatory Networks across Fly, Worm and Human
in preparation for Nature Biotechnology.
The term “in preparation” is common on academic websites, especially on sites of graduate students or postdocs who are eager to advertise forthcoming work, but “in preparation for” really requires some hubris. We have to wonder, why do Feizi et al. assume that peer review is just a formality in the journal Nature Biotechnology?
[Addendum 2/23: The authors posted a rebuttal to this blog post, which we replied to here.]

67 comments
Comments feed for this article
February 11, 2014 at 10:09 am
Manolis Kellis
We are unfortunately not the first (and probably won’t be the last) to be accused of deceit and fraud by Lior’s blog. We have tried to explain our work in the clearest and most transparent way possible, and stand by the importance of our results and wide applicability of our method.
We have heard very positive feedback from many other scientists using our software successfully in diverse applications, and others who have built upon it and extended it in their own work. We have also interacted with scientists who have diligently reproduced every figure of our paper.
We have described all pre- and post-processing steps of our algorithm in the supplement and in the code. We also show the performance of ND for various parameter settings. Unfortunately, the original supplement included an incorrect figure 4, which was changed in the updated version in August 26, 2013.
Regarding the availability of datasets, we only used public datasets that we have linked to from our paper and supplement.
Lastly, in our experience, partial correlation performed very poorly in practice, but we encourage Drs. Bray and Pachter to publish its performance in the DREAM5 benchmarks.
February 11, 2014 at 10:21 am
Lior Pachter
With all due respect, while I have critiqued many papers, this is the only time I have made an accusation of fraud. I am disappointed that your response does not address the specific questions 1 & 2 we ask in the box in the blog.
February 11, 2014 at 4:10 pm
Britney
Lior,
Manolis got very positive feedback, and as you should know by now, that’s what science is all about. Your nitpicking is quite pathetic.
Britney
February 15, 2014 at 6:35 am
harry
I have read this paper. I also felt confused about some details in this paper. Even though there are some faults you mentioned, there is not a perfect theory or algorithm.
I am interested in ND and did some experiments. I reproduced almost every figure in this paper. Due to lack of time, Figure S4 was not reproduced. It also took me some efforts to reproduced them. You know there are some details in experiments, not illustrated in paper. Feizi told me the details through emails patiently.
I think ND is a promising method. So I stand out for the truth.
February 11, 2014 at 8:31 pm
Manolis Kellis
Please find a lengthier response to allegations 1-6, and four appendices discussing: (A) robustness to linear scaling; (B) importance of eigenvalue scaling; (C) robustness to beta; (D) comparison to partial correlation; at http://compbio.mit.edu/nd/Response_to_Nonsense_Blog_Post.pdf
February 19, 2014 at 11:35 am
Anon
As asked on PubPeer, could you provide the names of some of the “many other scientists” who you “heard very positive feedback from”? You previously offered to do so by email, but have completely failed to follow up…
February 11, 2014 at 5:13 pm
Anon
The authors appear to be participating in the associated thread at PubPeer:
https://pubpeer.com/publications/542DE2FB103AE4942BC1972EBFF15E
Lior, you may wish to do so as well.
February 11, 2014 at 5:30 pm
AnotherAnon
>Manolis got very positive feedback, and as you should know by now, that’s what science is all about. Your nitpicking is quite pathetic.
On the contrary, scientific review is mostly about nitpicking, even when people insist on being lousy. Popularity contests are beside the point.
Plenty of instances of academic fraud have received positive feedback initially, take the ‘research’ in eugenics as a hyperbolic example.
February 11, 2014 at 5:54 pm
homolog.us
https://pubpeer.com/publications/542DE2FB103AE4942BC1972EBFF15E
“We have heard very positive feedback from many other scientists using our software successfully in diverse applications, and others who have built upon it and extended it in their own work. We have also interacted with scientists who have diligently reproduced every figure of our paper.”
I find it rather unusual that none of those ‘scientists’ came forward to claim $100 offered by Lior Pachter. It should be easy money !
February 11, 2014 at 8:32 pm
Manolis Kellis
Please find a lengthier response to allegations 1-6, and four appendices discussing: (A) robustness to linear scaling; (B) importance of eigenvalue scaling; (C) robustness to beta; (D) comparison to partial correlation; at http://compbio.mit.edu/nd/Response_to_Nonsense_Blog_Post.pdf
February 11, 2014 at 9:38 pm
homolog.us
Manolis, What you did with the supplementary figure is clearly fraudulent. You replaced Fig S4 in the updated version post-publication, which changed the qualitative nature of the figure and paper. However, there is no mention of that in the text that you provided related to revision (which I reproduce below).
“In the version of this file originally posted online, in equation (12) in Supplementary Note 1, the word “max” should have been “min.” The correct
formula was implemented in the source code. Clarification has been made to Supplementary Notes 1.1, 1.3, 1.6 and Supplementary Figure 4 about
the practical implementation of network deconvolution and parameter selection for application to the examples used in the paper. In Supplementary Data, in the file “ND.m” on line 55, the parameter delta should have been set to 1 – epsilon (that is, delta = 1 – 0.01), rather than 1. These errors have been corrected in this file and in the Supplementary Data zip file as of 26 August 2013.”
That means the reviewers saw one figure and the current version of the figure has a completely different version. , You may argue that qualitative change in Fig S4 has no effect on the main paper (or rather it is an uninformative figure). However, you were supposed to make that argument with the editor before replacing the figure. I would recommend that you withdraw and resubmit the paper, because the currently published paper is not peer-reviewed for the reasons stated above.
February 11, 2014 at 9:54 pm
homolog.us
The caption of Fig. S4 changed from –
” As it is illustrated in this figure, choosing β close to one (i.e., considering higher order indirect interactions) leads to best performance in all considered network deconvolution applications. Further, note that despite the social network application, considering higher order indirect interactions is important in gene regulatory inference and protein structural constraint inference applications.”
to –
” For regulatory network inference, we used β = 0.5, for protein contact maps, we used β = 0.99, and for co-authorship network, we used β = 0.95. Further, note that despite the social network application, considering higher order indirect interactions is important in gene regulatory inference and protein structural constraint inference applications.”
It is clearly a qualitative change and should have gone through peer-review, given that the supplement is part of peer-reviewed publication.
Your five-page rebuttal calls Lior Pachter’s post ‘sensational’, but as far as I can see, he merely stated a highly non-standard move made by you – namely making major changes to the supplementary figures without telling the editor. Do you ENCODE people really think that the rest of the world is full of idiots?
February 11, 2014 at 10:15 pm
AnotherAnotherAnon
Manolis: If you have indeed switched the supplementary figures post peer-review without the editor’s knowledge then it is unethical. This is going to attract much scrutiny to your past and future publications.
February 11, 2014 at 10:19 pm
UBN
Thanks to the blog authors for the diligence in uncovering another half-baked paper published in Nature. It’s a disgrace what the Nature family of journals has become; they are simply magazines that publish minimally peer-reviewed articles by those invited to be in the club. The authors’ rather weak and evasive reply was confirmatory of the gist of the blog post.
February 12, 2014 at 6:32 am
Edward Lear
Any journal can make a mistake. If the scientific community didn’t reflexively assume that a paper in a Nature journal is automatically worth 10x more than a paper in any other journal, it wouldn’t be such a problem.
I’m sure that PLoS One doesn’t check corrections to supplementary material any more carefully than Nature Biotech does….
February 12, 2014 at 6:40 am
Lior Pachter
Its a huge problem when an author deceives a journal and its readers, regardless of whether its Nature Biotechnology or PLoS One. Switching a figure means that the paper was effectively not peer reviewed, because what the reviewers saw was not correct. I don’t think its the job of any journal to police such activity as it goes without saying that authors should be honest and ethical. Having said that, if such activity is discovered, the penalty should be retraction of the paper (with option for resubmission and re-review). When Feizi et. al. switched that figure all of us, including the journal, became victims. If I was the editor of NBT who handled Feizi et al. I would be very very pissed off.
February 12, 2014 at 7:05 am
Edward Lear
Agreed. I would be too.
February 12, 2014 at 9:45 am
homolog.us
“The ‘Right Way’ and the ‘Wrong Way’ to Criticize Science”
http://www.homolog.us/blogs/blog/2014/02/12/lior-pachter-exposes/
I find it very telling that far fewer so-called scientists commenting in twitter are concerned about switching of figure than the ‘tone’ of Lior.
February 13, 2014 at 6:18 am
Manolis Kellis
We encourage readers of Lior’s blog to try ND out for themselves here:
http://compbio.mit.edu/nd/try_it_out.html
February 14, 2014 at 9:27 am
"It Is Not Hard to Peddle Incoherent Math to Biologists" - PharmaLeaders.com
[…] work from the lab of Manolis Kellis at MIT. (His two previous posts on these issues are here and here). I’m going to use a phrase that Pachter hears too often and say that I don’t have the […]
February 14, 2014 at 1:13 pm
Truth in advertising
I would say that the distinction between good marketing and misleading (or even fraudulent) claims is blurry and different people draw the line differently: From Feizi et al. point of view, they simply marketed their method well by casually simplifying certain details of their method in the main text of the paper (but providing code and supplementary material). Thus, they managed to “get a major paper”, rather than publishing this in a technical journal (where it probably belongs). Good for them.
On that note, I agree completely with Erik. Methods will have to stand the test of time. Every newly published method performs better than the competitors in the initial publication, because most authors use strawmen as comparisons. That is why we have independent competitions for these things (such as DREAM) and we shall see how well the method will perform there.
February 14, 2014 at 7:21 pm
A Senior Scientist in CB
Manolis Kellis: I believe you need some time to think about yourself and the way you do research. Just chasing the latest data and trying by all means to publish will not be good for yourself, a scientist. I believe that you need to sit in some maths and algorithm classes. It’s good that Lior was brave enough to stand up and criticize. It’s good not only for you, but also for the computational biology field.
February 15, 2014 at 8:41 am
"It Is Not Hard to Peddle Incoherent Math to Biologists" – JumpSeek
[…] work from the lab of Manolis Kellis at MIT. (His two previous posts on these issues are here and here). I’m going to use a phrase that Pachter hears too often and say that I don’t have the […]
February 15, 2014 at 8:48 am
Weekend reads: MIT professor accused of fraud, biologist who retracted paper suspended, and more | Retraction Watch
[…] of California, Berkeley’s Lior Pachter rips apart a pair of papers by MIT’s Manolis Kellis, accusing Kellis of fraud, and Kellis […]
February 15, 2014 at 9:38 am
Weekend reads: MIT professor accused of fraud, biologist who retracted paper suspended, and more – Nouvelles et satellite scientifique
[…] of California, Berkeley’s Lior Pachter rips apart a pair of papers by MIT’s Manolis Kellis, accusing Kellis of fraud, and Kellis […]
February 15, 2014 at 12:20 pm
Reproducility
Hi Lior:
You did the scientific community a service and I thank you for the time and effort that you and your student spent. I do think this goes to highlight the worrisome trend (the authors of the two papers are hardly alone in this) to try to publish a Nature/Science/Cell paper no matter what. However, hyping results is something that many/most people do to varying degrees. I am not convinced that Feizi et al. crossed the line to outright fraud.
[…] we have been unable to replicate the results of the paper. […]
This is a very serious accusation, as it implies fraud. I think it would be another service to the community if you could post online the results that you get when running the authors code with the parameters they specify on the matrix provided, so that everyone can compare (If I had a day to spare I’d do it myself). Reproducibility (or lack thereof) should be something decidedly easy to establish in compbio.
Sincerely, a concerned fellow scientist.
February 20, 2014 at 1:30 am
Rififi chez les bioinformaticiens : peut-on tout critiquer sur tous les tons ? | Tout se passe comme si
[…] dernière. Dans une série de trois billets (The network nonsense of Albert-László Barabási ; The network nonsense of Manolis Kellis ; Why I read the network nonsense papers ; plus une explication de texte finale : Number […]
February 25, 2014 at 3:10 pm
Lior Pachter’s Blog | Xiaole Shirley Liu's Blog Site
[…] some very provocative blog articles on Lior Pachter’blog attacking Barabasi and Kellis’s recent network biology papers. Lior is probably a little harsh in his tone, but the irregularities […]
February 28, 2014 at 9:43 am
HN
Highlight track on April 4th: http://www.compbio.cmu.edu/recomb/recomb14%20tentative%20program.pdf
March 1, 2014 at 12:01 am
village-idiot
I must say, this nerd war between you and Manoli is probably the most exciting development in comp bio in recent memory. Although, I would request that you focus your energies on Barbasi, who publishes hilarious shit like “The Product Space Conditions the Development of Nations” in journals like Science….the dude hangs out at the World Economic Forum (according to wikipedia) and has an H-index of 103 (WTF!!!?!?!?!).
In all seriousness though, as a computational biologist, I would say that 99.9% of the papers in our field do not advance biology. It’s great that you have called out these two papers, but I think it’s safe to say that they were going to have zero impact on biology anyway. There is an obsession with running large scale experiments which produce a bunch of meaningless papers — almost all of them have a pretty clustergram picture followed by a hairball of nodes and connections, and somehow that’s supposed to be very impressive and useful in designing follow-up experiments.
March 9, 2014 at 9:13 pm
GradStudent
Definitely many interesting points here. But dude, CHILL OUT!! How does angrily attacking a graduate student help your cause in any particular way?
I don’t see why you had to go so far as to make fun of Feizi’s “in preparation for”. One may in principle conduct his or her study “in preparation for” receiving the Nobel Prize one day. I don’t see any problem of saying that. Making fun of Feizi’s website just sounds mean and cheap.
I get it, you had a rough patch with Kellis years ago. But what did Feizi do to piss you off (and for that matter Kellis’ other students)? The poor fellow has to get a job some day. At least you could have stated your points nicely and in a constructive way without the mockeries.
I feel bad for your students, and the students of every other prof that you don’t seem to like.
– a grad student (life is a bit rough for us, in case you don’t remember any more)
March 11, 2014 at 4:42 am
Lior Pachter
Dear GradStudent,
Regarding the specific matter of Feizi’s “in preparation for (Nature Biotechnology)” language for multiple papers on his publication page, I believe it is very fair for me to point out and readers can draw their own conclusions about whether it is relevant to the points I made in the blog post (note: he immediately removed the language after my blog post). As for his future job prospects, I certainly believe they should depend on (the quality of) his publication record.
I cannot respond meaningfully to your other attacks unless you de-anonymize yourself, except to say that I have never had “a rough patch” with Kellis as you claim, and I am not sure on what basis you make that claim.
Sincerely,
Lior
March 11, 2014 at 9:57 pm
Pinko Punko
GradStudent- I can’t speak for anyone else, but I did not read the comment about “in Preparation for” as making fun. This is common enough behavior currently, but in some circles that can be seen as bad form. I don’t think Lior needed to go there because conflating possible motivations of Kellis with what could have just been an frank statement of a grad student “we’re writing this and we’re sending it to NB” seems unnecessary. The rest of your comment seems to negate your statement that there is something interesting here- because you move on to say that you “get it” claiming this must all be some sort of conflict of personalities.
March 12, 2014 at 7:15 am
Anon
GradStudent, I was with you until “the poor fellow has to get a job some day”. Everyone has to get a job! I was a grad student at one point too and I certainly remember how rough life was. It definitely doesn’t help other grad students if one person gets an advantage unfairly. Also, regarding “what did Feizi do to piss you off”, I think this blog described that in gory detail already, whether or not you think the actions were fraudulent.
March 11, 2014 at 7:38 am
Friday 14th: network inference and suspicious methods for that | Helsinki Statistics Discussion Club
[…] https://liorpachter.wordpress.com/2014/02/11/the-network-nonsense-of-manolis-kellis/ […]
March 11, 2014 at 11:44 am
Anonymous
Lior,
You mention above that as the scaling goes to zero in (3), $G_{dir}$ converges to $G_{obs}$. But, shouldn’t $G_{dir}$ approach an identity matrix in the limit $\gamma\rightarrow 0$?
Thanks.
Spectator
March 14, 2014 at 10:50 am
Lior Pachter
With a scaling factor gamma, we have that G_dir = gammaG_obs(I-gammaG_obs)^{-1}. (I-gammaG)^{-1} is approximately I+gammaG_obs, and therefore G_dir is approximately gammaG_obs + gamma^2G_{obs}^2. As gamma approaches zero the latter term goes to zero, so that G_dir is approximately cG_{obs}. This converges to the zero matrix, but for a nonzero gamma, it scales back to G_{obs} by multiplying by 1/gamma. That is what I mean by the claim that as gamma->0 the procedure converges to G_dir -> G_obs.
March 14, 2014 at 5:33 pm
Anonymous
Thanks for your reply. Using the notation in (3), one actually gets G_dir = (G_obs/gamma)(I-G_obs/gamma)^{-1}. In this notation, as gamma goes to 0, G_dir approaches identity. Anyway, just a minor point. Thanks.
March 14, 2014 at 9:57 pm
Lior Pachter
Sorry for the confusion. By “the scaling” I mean 1/gamma in the post, and of course in my comment I should have stuck to that instead of referring to the inverse. The main thing is I think we are now on the same page 🙂 Thanks!
March 12, 2014 at 9:49 pm
Anti-MIT-EECS
Wow, I am surprised that after all of these fantastic posts by Lior, MIT EECS didn’t even take a single action against their “Full Professor” Manolis Kellis =))
https://www.eecs.mit.edu//news-events/media/kellis-stultz-weiss-and-lu-receive-faculty-promotions
March 26, 2014 at 3:27 pm
anonymous reader
I’m guessing MIT EECS doesn’t even know about this.
Even if they did, what would they do?
He’s made sure to work with some big name people, so he’s likely to be politically well connected…
March 18, 2014 at 4:01 pm
Reproducibility vs. Usability | Bits of DNA
[…] papers. Here I describe a case study in reproducibility and usability, that emerged from a previous post I wrote about the […]
April 20, 2014 at 7:51 pm
Does researching casual marijuana use cause brain abnormalities? | Drug Policy Debate Radar
[…] is quite possibly the worst paper I’ve read all year (as some of my previous blog posts show I am saying something with this statement). Here is a breakdown of some of the issues with […]
June 16, 2014 at 2:58 am
Hedi Hegyi
I think the original idea of network deconvolution is brilliant. I suspect the gene regulatory networks are a lot more complex than they seem from their correlation matrices. Also, while in the protein-protein interactions and in the coauthorship networks there are only positive or zero elements (i.e. two proteins either interact or not, two scientists either published a paper together or not), as Lior points out himself, there is the unfortunate fact that some genes negatively regulate other genes’ expression, that might be hard to capture with this model. The situation is akin to introducing, say, “negative publications”, e.g. this feud between Lior and Manolis could be considered one 🙂 Try to recalculate network deconvolution of co-authorship after introducing such negative elements!
August 9, 2014 at 11:40 pm
helloworld
Hey there, I think your blog might be having browser compatibility issues.
When I look at your website in Safari, it looks fine but
when opening in Internet Explorer, it has some overlapping.
I just wanted to give you a quick heads up! Other then that, great blog!
September 2, 2014 at 12:33 pm
Crossroads (ii) – Is It Too Late to Acknowledge that Systems Biology and GWAS Failed? « Homolog.us – Bioinformatics
[…] of ‘omics’ terms and inflated claims about the success of computer algorithms (check The network nonsense of Manolis Kellis and related posts in Lior Pachter’s blog), but hardly any real contribution was made, as Dr. […]
September 25, 2014 at 8:45 pm
Random Variable
Lior
I know some people who won genius awards with unremarkable garbage work, some who became members of NAS, NAE, Royal Society, etc. with making noise, and playing politics. Modern academia is about marketing and making noise–whether we like it or not.
I think you should funnel your anger at commercialization of science and education. Attacking these guys (although I think you have some valid points) is not going to help much. They are just products of the system.
November 24, 2014 at 10:19 am
Random Foo
Random Variable,
You talk about marketing and politics. Even if we were to agree with that ideaology, Manolis is very disagreeable.
December 30, 2014 at 9:57 am
The two cultures of mathematics and biology | Bits of DNA
[…] between the worlds of mathematics and molecular biology in my own institution. I’ve also seen the consequences of the separation of the two cultures. To illustrate how far apart I’ve made a list of […]
February 20, 2015 at 9:26 am
The Conspiracy of Epigenome? « Homolog.us – Bioinformatics
[…] leader of the epigenome project, was accused of fraud by Berkeley mathematician Lior Pachter (check here and here). In the scientific world, a fraud allegation is far more serious than someone’s […]
April 8, 2015 at 2:35 pm
Anonymous
A corrigendum was published yesterday in NatBiotech http://www.nature.com/nbt/journal/v33/n4/full/nbt0415-424.html claiming the method performs even better than previously reported …
April 9, 2015 at 4:54 pm
Lior Pachter
I’m pleased to see that Nature Biotechnology is re-evaluating editorial oversight of computational biology papers, and applaud the editors for acknowledging the importance of reproducibility and usability. Even more important, is their recognition that “rigorous peer review [of computational biology papers] is likely to change”. Both the Barzel-Barabasi and Feizi et al. papers demonstrate that computational biology review requires critical evaluation not only of purported biological results, but also of the underlying statistics and mathematics.
As a result of this blog post and others, I have received lots of feedback from many students, colleagues and editors on the challenges and opportunities of computational biology and I thought that in the context of the Nature Biotechnology editorial I would share some of the ideas that have been floated (in no particular order order):
A lot of biology research, and certainly computational biology research, is now collaborative. Yet referees are frequently asked to review papers in isolation of each other (eLife is an exception to this). It seems that collaborative reviewing would help in the evaluation of interdisciplinary work, and also facilitate the bridging of gaps between fields.
The review of papers in mathematics, statistics and computer science is frequently a collaborative rather than an adversarial endeavor. The review of a paper is considered a privilege, and reviewers may go so far as to (anonymously) improve theorems or proofs in submitted papers. I believe this happens because sub-fields are small, people generally know each other (from conferences and via collaboration), and reviewers acknowledge hard work with the favor of thorough review. Computational biology however is a fragmented field, spanning multiple disciplines and communities, and there is no coherent single community. In particular, there is a disconnect between “computational” conferences such as RECOMB or ISMB, and biology conferences. Fostering a culture of collaborative review in computational biology will require journal editors, conference organizers, and leading scientists (both biologists and computational scientists) to make overt efforts to overcome cultural barriers and to embrace each others strengths. Building a strong cohesive community of scholars in computational biology will also help editors and others to identify experts.
Good reviews of biology and mathematics must, fundamentally, be very different. A critical view of biology research involves assessment of whether appropriate controls have been performed, whether results are based on sufficient evidence, and whether obvious follow-up questions have been explored (to name a few things). One thing biology reviewers do not do is try, on their own, to replicate experiments. This is simply not feasible- it would be too expensive and too time consuming. Replication happens after the fact (of publication). To some extent, the review of biology papers requires trust and faith, and for this reason the lab the work originates from factors into the belief of reviews in the work. In mathematics, the opposite is true. Proper review of research always involves the checking (and therefore replication) of results. For example, in mathematics or statistics, each and every step of a proof is carefully checked by reviewers. This is different than the kind of replication Nature Biotechnology is discussing in its editorial, but it is equally important. Mathematics review therefore requires more time, and journals will have to adjust reviewing, and possibly publication procedures accordingly.
Computational biology research is exciting because of its interdisciplinary nature, but also challenging for that very reason. It requires facility in multiple areas, and the bar is very high. Researchers are asked not only to develop new methods, but also to demonstrate their applicability on “real data”, and to furthermore use them to discover new biology. Policies enforcing reproducibility and usability are important and necessary, but also place additional burdens on researchers that are not required or enforced in other fields. I therefore think it is important for journals to think not only punitively, but also positively. I’d like to see journals advocate for and reward computational biologists for good work. This could be done via changes in policies on authorship (e.g. when co-first authors are starred as equal contributors, journals could alternate display of first names in online editions), promotion of editorials and reviews by computational biologists, and acceptance of “pure” methods papers as viable and important in their own right.
These are just a few thoughts– hopefully the Nature Biotechnology editorial will spur more discussion on the issues surrounding publication and review of computational biology and lead to improvements in the quality of published work in the field.
April 9, 2015 at 7:49 pm
Yu Xue
Hi Lior, three days ago I just saw the criticisms on the Barabasi’s NBT paper, and the authors’ response. Now it’s clearly to me that you told the truth: The network nonsense was reported. I will write a blog on it, in Chinese. – Yu
May 31, 2015 at 2:04 pm
Criticize me like a scientist – JEFworks
[…] in tweets, blogs, or publications, words can get thrown. Scientifically valid but often colored with not the most […]
June 11, 2015 at 5:49 am
In which I’m pretty sure I disagree with Lior Pachter and try to figure out why | beanbag genomics
[…] sucking up resources like grant money, then you should Take. Them. Down. By any means necessary. Accuse them of fraud. Call them innumerate. Mock their minor errors. Is your colleague a male that asks lots of […]
June 11, 2015 at 6:54 am
Lior Pachter
Dear Joe,
You make many claims in your blog post that are completely unfounded and presumptuous. You say that “I mostly dislike people who don’t use Cufflinks”. Could you please point to any evidence for this assertion? Nothing could be farther from the truth and I believe that my actions and statements on RNA-Seq and RNA-Seq software speak very clearly for themselves. I mean, do you think I hate myself? You say that I wrote ” The most embarrassing citation ever?” to mock people. On what basis do you say that? I made it very clear in my post that my concern was specifically with the practice of using software that is unpublished, and then citing it via websites. To wit, when the authors wrote to me personally to let me know they would ask the journal to “fix” the website link to the Flux Capacitor website, I replied that this was not the right thing to do, and that instead they should use published software (I personally believe even BiorXiv or arXiv is a publication; for Flux Capacitor there is literally nothing). Was their action embarrassing? Very much so. I highlighted it because it is relevant in the context of a general disdain for computational biology methods in biology papers. You say that I see members of the GTEx consortium as my “adversaries”. This could not be farther from the truth. A number of GTEx consortium members are my colleagues/collaborators and people I very much respect. You just made that up. You write that my objective is to “Take Them Down” (I assume your “them” refers to Manolis Kellis). It is of course your prerogative to presume whatever you wish, but you could have just asked me what my objective is instead. You mock my accusation of fraud (I assume you are referring to my post on the Network Nonsense of Manolis Kellis) but in fact, it is a very very serious accusation that I made carefully and after much consideration. I don’t think it, and the other criticisms I’ve posted constitute “minor errors”. If you think so then all I can suggest is please read the posts. Otherwise what you have written is just
slanderlibel.Lior
June 11, 2015 at 11:55 pm
MaybeNextTime
Lior,
Interesting that you don’t actually say what the objective of repeatedly targeting Manolis Kellis’ work *is*. If the point of your blog is to highlight examples of science that you find substandard, why the relentless focus on the work of one person? Surely there are many other examples around that you could direct your energies towards? Also – if your motives are as high-minded as you claim – how do you not understand that the single-minded pursuit (some might say obsession) with the work of one person seriously undermines any claim you have to legitimacy. For one thing, it helps perpetuate that “devil / saint dichotomy” that you’ve said you’re trying to get away from. How does vilification of one person’s work achieve this, exactly? For another, repeated targeting of the same person looks suspiciously like a grubby personal vendetta, not some noble quest for truth. I’m not saying it *is*, but I don’t think it does you any favours.
June 12, 2015 at 5:39 am
Lior Pachter
First of all, it is a complete mischaracterization of my blog to refer to it as a “single-minded pursuit” of Manolis Kellis. I’ve put up >50 posts which collectively mention many papers (probably >100?), and by my count 5 papers on which he is a coauthor appear among them. Please note that my blog focuses on computational biology and considering his publication rate is among the highest in computational biology making him one of the most prolific researchers of our time (e.g. he has published >16 papers according to Google Scholar so far in *2015*) the fact that more than one of his papers has been mentioned is hardly surprising. Also he is involved in almost all high profile projects in computational biology: ENCODE/modENCODE/GTEx/Epigenome Roadmap, etc. etc. And, just to make a technical point, when people refer to “his work” they are taking a very broad perspective. My post on the p-value prize referred specifically to a paper on which he was first author and Eric Lander last author. In this post he is last author. On the GTEx paper he is somewhere in the middle. How are these all “his work” or “his papers”?
You asked about my objectives and I’m happy to explain them as I have many times on various pages of this blog, including in a lengthy recent comment above. In no particular order: I am advocating for a shift in the view of computational biology as a service thereby appropriately relegated to the status of supplementary material in papers, to an understanding that it is a legitimate research enterprise. I am advocating for higher standards of work in computational biology papers, for example that they be reproducible and usable, and also for higher standards in their review. I am advocating for a change in the paradigm of journal pre-publication peer-review which is failing authors and science and is particularly ill-suited to computational biology. I am advocating for returning vision to the blind-eye which allows for fraud in computational biology of the nefarious nature I’ve described in this post. I am advocating for a culture of post-publication peer review. My objective(s) are that all these things happen.
Hope that helps.
June 15, 2015 at 6:47 am
cbouyio
Reblogged this on CBouyio.
May 30, 2016 at 2:06 pm
Paul
I am strongly supporting Lior for writing this blog. As a applied mathematician who is using solid mathematics to help people solve challenging problems, I am in fact very angry and surprised about the fact that these people from MIT used elementary school mathematics, without any proof or quasi-proof of optimality, to solve a trivial mathematical problem they proposed [which may have nothing to do with the practical question people care about]. Lior didn’t spend too much time questioning their assumption since other parts already deserve lots of criticism, but I would like to say that the extremely trivial and naive assumption makes this paper completely meaningless. I can think of a more extremal example: suppose I would like to recover ground true A from observation B. I first assume that B = A+3. Then, I define \hat{A} = B-3 as my recovered ground truth, and claim it is optimal. The very obvious bug of this approach is that one can never justify that why B = A+3! Although this example is overly simplified, I think it is precisely what is underlying this paper. It is really a shame on the authors and on the journal. I suggest anyone who wishes to apply mathematics to practice study this paper to remind himself/herself of what bad research is like.
January 18, 2017 at 1:45 pm
A citation is not a citation is not a citation | Bits of DNA
[…] and Koeppl 2016 who show, with specific examples, that the scaling parameter we criticized in Bray & Pachter 2013 is indeed […]
January 31, 2017 at 8:06 am
This Week in Data Reading – Data for Breakfast
[…] figures in this scientific field. (Note that Pachter’s critiques can be harsh as, for example here or here; be sure to read the comments for criticism on the critique itself.) If you write or read […]
August 17, 2017 at 2:17 pm
Jonathan
Dear Professor,
Thank you for sharing this! As an undergrad doing research in computational biology, I learned a lot about best practices from this.
I just have one question: are the two contentions:
1.the method used to obtain the results in the paper is completely different than the idealized version sold in the main text of the paper and
3. the authors appear to have deliberately tried to hide the existence of the parameters.
necessarily indefensible, by themselves? (I wholeheartedly agree with the problems of #2 and 4-6)
When writing the main manuscript or giving a presentation on a complex algorithm, isn’t it often important to simplify and just show the main idea so that the audience can wrap their heads around the general approach? Otherwise, it might be hard to understand if you explain every necessary details.
In this case, I realize that it raises drastic problems in terms of identifiability of the solution, but in general, would you think it’s okay to simplify in the manuscript and give full detail in the supplement?
March 9, 2018 at 3:36 pm
rishabh
Hi Lior, your write-up is really interesting. What do you think of multi-task learning for network inference, I found a few examples online:
Click to access 279224.full.pdf
Click to access 279224.full.pdf
February 11, 2019 at 4:30 pm
Nonsense methods tend to produce nonsense results | Bits of DNA
[…] years ago on this day, Nicolas Bray and I wrote a blog post on The network nonsense of Manolis Kellis in which we described the paper Feizi et al. 2013 from the Kellis lab as dishonest and […]
May 26, 2020 at 11:04 pm
Monitoring progress in translational bioinformatics - PLOS Biologue
[…] is mandatory reading for anyone interested in translational genomics. Next, I highlighted the blog of colleague Lior Pachter, as he engaged in an entertaining (and informative) polemic about network […]
November 17, 2020 at 12:28 pm
Mathematical analysis of “mathematical analysis” of a vitamin D COVID-19 trial | Bits of DNA
[…] matched will be required go show a definitive answer”. In a follow-up paper, Jungreis and Kellis re-examine this so-called “Córdoba study” and argue that the authors of the study have […]